**節(jié) 細胞培養(yǎng)在分子生物學中應用
分子生物學是在生物化學、遺傳學、免疫學、微生物學、細胞生物學、生物物理學等學科結合的基礎上,經過相互雜交、相互滲透融合發(fā)展起來的一門新興學科,它從分子水平上研究生命現象的本質以及活動規(guī)律,以達到造福人類的目的,主要側重于研究基因的結構和功能、分子間信號傳遞和調控。
基因在細胞中的表達在時間和空間上具有高度的特異性;細胞的運動、遷徙、粘著、分化的控制機制需要從分子水平上闡明;腫瘤的發(fā)生和發(fā)展的機制有待于深入研究;人類基因組計劃已于2000年6月完成人類基因組草圖,科學家發(fā)現人類基因數目約為3萬個左右,僅比果蠅多2萬個,遠少于原先10萬個基因的估計。2003年4月14日,G際人類基因組測序組織正式對外宣布:美、英、日、法、德和中G科學家經過13年努力共同繪制完成了人類基因組序列圖,人類基因組計劃的所有目標均已實現。目前基因功能比較清楚的有10 000條左右,還有大量的基因功能不明;基因的功能需要以細胞為載體,細胞為這些研究提供了研究的對象和材料。
鄂征等將以研究體外培養(yǎng)的細胞為主要研究對象的分子生物學技術,稱為分子細胞學技
術。細胞是由多種生物大分子如核酸、蛋白質、類脂等組成并由它們行使各種功能活動,只有通過對它們的研究,才能了解細胞的基本活動規(guī)律。
對其的研究技術可以分為三類,一是分離提取研究;二是基因導入研究;三是原位檢測研究。
一、分離提取研究
根據分離物質的種類分為DNA提取、RNA提取、蛋白質提取。
1.DNA提取 在分子生物學研究中,DNA是這些技術應用的主要對象,所以從真核細胞中分離DNA是分子生物學研究中很重要的基本技術,DNA樣品的質量直接關系到實驗成功與否。真核細胞95%的DNA存在于細胞核中,DNA與蛋白質結合在一起,構成染色質的**結構,另有5%存在于細胞器(線粒體)中。提取DNA的方法比較多,但基本原理是一樣的,即去除與核酸結合的蛋白質以及多糖、脂類生物大分子,去除其他不必要的核酸分子,沉淀DNA,去除鹽類、有機溶劑等雜質,**后純化核酸。
方法一:酚抽提法
【材料】
磷酸鹽緩沖液(PBS,pH7.4),DNA抽提緩沖液,10mmol/L Tris-Cl(pH8.0),0.1mol/L ED-TA(pH8.0),20μg/ml RNase A,0.5%SDS,蛋白酶K 20mg/ml,酚(Tris-C1 pH8.0,飽和),NH4Ac 10mol/L,TE(10mmol/Tris-Cl(pH8.0),1mmol/L EDTA(pH8.0)),無水乙醇。
【操作過程】
(1)收集細胞,對于懸浮培養(yǎng)的細胞可以直接經1500g離心,用PBS洗滌一次,再次離心去上清;對于貼壁培養(yǎng)的細胞,用胰酶消化后再離心收集,用PBS液洗滌一次后,再次離心,棄上清,收集細胞。
(2)在有細胞沉淀的離心管中加入DNA抽提緩沖液l0ml,37℃ 1小時。
(3)加入50μl 20mg/ml的蛋白酶K,50℃保溫3小時。
(4)加入等體積的酚,上下顛倒混勻,直**水相與酚相混勻成乳狀液。
(5)室溫,5000g離心15分鐘,將水相轉移到另一離心管中,重復酚抽提二次。
(6)轉移水相,加入0.2倍體積的10mol/L NH4Ac,2倍體積的無水乙醇混勻,可立即看到乳白色絲狀沉淀,棄去上清,立刻用70%乙醇漂洗沉淀。
(7)室溫,5000g離心5分鐘,棄上清,室溫讓乙醇揮發(fā)干凈。溶于適量的TE中。
(8)測定DNA的含量和純度。
【注意事項】
(1)此法可以用于提取5×107個細胞,可得產量為200μg。
(2)高分子量DNA提取過程中,取上層DNA,往往會牽動水相與酚相的界面蛋白質層,可以將Tip頭剪去一段,這樣**的口徑增大,緩慢吸取水相。
(3)在DNA抽提緩沖液中加入高濃度的RNase A,可以省去DNA沉淀后再次用RNaseA消化殘留的RNA。
(4)此方法可分離100~200kb左右的基因組DNA,適用于λ噬菌體作為載體的基因組文庫的構建、脈沖場電泳、酶切分析、Southern雜交、PCR檢測等實驗。
方法二:甲酰胺解聚法
【材料】
DNA抽提緩沖液:10 mmol/L Tris-C1(pH8.0),0.1mol/L EDTA(pH8.0),20μg/ml RNase A,0.5%SDS。蛋白酶K 20mg/ml。變性緩沖液:80%去離子甲酰胺,0.8mol/L NaCl,20mmol/Tris-C1(pH8.0)。透析液1:0.1mol/L NaCl,20mmol/L Tris-Cl(pH8.0),l0mmol/L EDTA(pH8.0)。透析液2:10mmol/L NaCl,l0mmol/L Tris-Cl(pH8.0),0.5mmol/L EDTA(pH8.0)。
【操作程序】
(1)細胞的收集及蛋白酶K消化同酚抽提法。
(2)當蛋白酶K消化后,將溶液冷卻**0℃,加入10ml變性液緩沖液,放置15℃過夜。
(3)將液體轉移**1個火棉膠袋中,先用透析液1透析,每次1L,換液4次;然后用透析液2透析,換液6次,每次700ml。
(4)直到DNA溶液中A260/280的值稍大于1.75,若低于1.75則需繼續(xù)透析。
(5)根據0D260計算DNA的含量,用脈沖場電泳分析DNA分子量的大小。#p#分頁標題#e#
【注意事項】
(1)此法是裂解細胞和消化蛋白后,利用高濃度的甲酰胺解聚蛋白質與DNA的結合,然后透析處理DNA樣品,由于提取過程中操作步驟少,對DNA的機械損傷比較小,DNA分子量可達200kb左右。其用途同酚抽提法。
(2)對于5×107個細胞,使用本法,**后的溶液體積為20ml,濃度為5~10μg/ml,在此濃度下,其溶液仍然很粘稠,說明DNA的分子量比較大。
方法三:Trlzol法
此法利用DNA和RNA在溶液中的分布不同將其分開,RNA分布在水相中,而DNA分布在有機相中,用乙醇將DNA沉淀下來。
【材料】
Trizol試劑,含0.1mol/L檸檬酸鈉的10%乙醇,無水乙醇,8mmol/L NaOH。
【操作程序】
(1)收獲細胞5×106個,方法同前。
(2)在含有細胞團的Eppendorf管中加入1ml Trizol試劑,振蕩混勻,室溫放置5分鐘。
(3)加氯仿0.2ml,振蕩混勻,室溫放置2分鐘。
(4)4℃,12 000g離心15分鐘,小心吸取上層水相,移人另一Eppendorf管中(用于RNA 提取)。
(5)在有機相中加入0.3ml的無水乙醇,混勻,室溫放置2~3分鐘。
(6)4℃,10 000g離心5分鐘。
(7)吸去上清(用于蛋白質的提取),用1ml含0.1mol/L檸檬酸鈉的10%乙醇漂洗DNA。
沉淀2次。在每次漂洗過程中,使DNA沉淀在溶液中保持30分鐘,其間不斷用手指振蕩。**后4℃,10 000g離心5分鐘收集DNA沉淀。
(8)用75%乙醇洗滌沉淀1次。
(9)空氣中干燥DNA沉淀(不要作真空干燥,也不要過度干燥,否則很難溶),用600μl 8mmol/L NaOH溶解DNA沉淀。
(10)此時,DNA沉淀中可能含有一些膠狀不溶物,可以將其在4℃,12 000g以上離心10分鐘,取上清。
2.RNA提取 在研究基因表達及其調控、cDNA合成、cDNA文庫的構建過程中,RNA的提取是必不可缺少的一步。在哺乳動物細胞中,平均每個細胞含有10-5μg RNA。對于培養(yǎng)的細胞而言,lg細胞相當于lml壓積的細胞,大約108個左右。在細胞質總RNA中,rRNA占80%~85%,mRNA占1% ~ 5%,余下的為tRNA和核內小分子RNA。其中mRNA分子種類繁多,分于量大小不一,在細胞內含量少,它是蛋白質合成的直接模板,是分子生物學中基因表達研究的主要對象。jue大多數mRNA分子(除血紅蛋白、干擾素和有些組蛋白mRNA以外)的3’端存在著20 ~ 250個多聚腺苷酸(PolyA)。利用此特征,可很方便地從總RNA中,用寡聚(dT)親合層析柱分離出mRNA。
RNA的分離關鍵因素是盡量減少RNA酶的污染。RNA酶是一類生物活性非常穩(wěn)定的酶,除細胞內RNase以外,環(huán)境中的灰塵、各種實驗器皿和試劑、人體的汗液以及唾液中均存在RNase。這類酶耐熱、耐酸、耐堿,蛋白質變性可使之暫時性失活,但在變性劑去除后,又可恢復活性。所以在實驗操作的過程中,應盡量減少RNase污染的機會。在整個操作中,在一個潔凈的環(huán)境中,帶手套和口罩,所用的器皿、水和試劑需用DEPC(焦碳酸二乙酯,是很強的RNase抑制劑)處理過。
玻璃器皿的處理。在常規(guī)洗凈后,應用0.1%DEPC浸泡2小時以上,再用雙蒸水漂洗幾次,高壓消毒去除DEPC,然后250℃烘烤4小時以上。塑料器材應用一次性、新的,如Eppendoff管、Tip頭等,在使用前高壓消毒,嚴格一些可以用0.1%DEPC浸泡過液,高壓消毒。所用試劑應加DEPC**0.05% ~ 0.1%,室溫過液,然后高壓處理。但要注意,DEPC易與Tris反應,所以在配制Tris時,應用DEPC處理過的水配制,**后再一次高壓消毒。
方法一:異硫氰酸胍、酚、氯仿一步法
此方法將已知**強的RNase抑制型異硫氰酸胍、9-巰基乙醇和去污劑N-十二烷基肌氨酸鈉聯合使用,抑制RNA的降解,增強了對核蛋白體復合物的解離,提高了RNA的產率。RNA選擇性地進入無DNA和無蛋白質的水相,容易被異丙醇沉淀濃縮,比較適合于大量或少量的細胞RNA提取。用此種方法提取的RNA可用于Northern雜交、cDNA的合成、體外翻譯、RT-PCR等。
【材料】
(1) DEPC處理水。
(2) 0.75mol/L檸檬酸鈉:稱取檸檬酸鈉(Na3C6H7·2H2O)22g,溶于70ml水中,以濃HCl調pH**7.0**100ml,高壓滅菌。
(3) 10%十二烷基肌氨酸鈉(sodium laurylsarcosine):稱取10g十二烷基肌氨酸鈉,溶于90ml水中,65℃助溶,定容**100ml。
(4) GSS緩沖液:用293ml 水溶解250g異硫氰酸胍,加入17.6ml 0.75mol/L檸檬酸鈉,26.4 ml 10%十二烷基肌氨酸鈉,濾過除菌。
(5) 變性緩沖液:在用時mi GSS緩沖液加入100社琉基乙醇
(6) 酚(水飽和);在100ml重蒸酚中,加入30mlDEPC處理水,磁珠攪拌混勻,直**酚相與水相形成明顯的界面。
(7)2mol/L NaAc(pH4.0)。
(8)氯仿:異戊醇(49 : 1)。
(9)PBS緩沖液(DEPC處理水配制)。
【操作程序】
(1) 收集細胞5×106個,方法同DNA提取。
(2) 在含有細胞團的Eppendoff管中加入0.5 ml變性緩沖液,振蕩混勻。
(3) 加入50μl 2mol/L NaAc,混勻后,加入0.5ml水飽和酚,再加入100μl氯仿/異戊醇,振蕩混勻后置于冰上15分鐘。
(4) 4℃12 000g離心15分鐘。
(5) 小心吸取上清,轉移**另一Eppendoff管中,加等體積的異丙醇,混勻后,置于-20℃**少一小時。在吸取上清時,不能接觸水相與酚相的界面,此界面為蛋白質層,酚相中主要為DNA。#p#分頁標題#e#
(6) 4℃,12 000g離心15分鐘,棄上清,加入150μl變性液,振蕩溶解沉淀(沉淀中含有RNA)。加入150μl異丙醇,置于-20℃,1小時。
(7) 4℃,12 000g離心15分鐘,棄上清(如不立即用,可以將此置于-70℃,長期保存)。
(8) 冰浴冷的75%乙醇漂洗沉淀;4℃,12 000g離心5分鐘,棄上清(小心不要將沉淀倒掉)。
(9) 真空干燥.去除樣品中的乙醇(但不宜完全干燥,否則沉淀難以溶解)。
(10) 用適量的DEPC處理水溶解沉淀,測定OD260、280、230,計算RNA的含量和純度,同時取大約150ng總RNA電泳檢測完整性。可以用非變性的瓊脂糖凝膠,EB染色后,可以見到有3條明顯RNA帶,分子量大的兩個分別為28S和18S,帶的亮度比為2︰1,如果小于此值,說明RNA存在著降解;如用甲醛變性膠電泳,則需要比較大的上樣量。
【注意事項】
(1) RNA的含量計算:X(μg/μl)=OD260×40×稀釋倍數/1000,OD260加為波長260nm時吸光度值。
(2) 通常純的RNA,OD260/OD280 =2.0,由于所用的標本不同,此數值在1.7 ~ 2.0范圍內波動。若低于此值說明有蛋白質污染,此樣本應再經酚/氯仿/異戊醇抽提一次。有時樣本此值大于2.0,如重復測定無誤,并不表明純度有問題。RNA樣品OD260/OD230常大于2.0,低于此值表示有異硫氰酸胍污染,此時樣品需經異丙醇沉淀,以除去小分子胍類的污染。
方法二:Trizol 試劑提取
Trizol試劑提取RNA的方法是由Invitrogen公司推出的新產品,其操作簡單、方便,在1小時即可完成,所制備的RNA可以用于Northern雜交和cDNA的合成。此種方法,還有一個優(yōu)點,它可以同時分離DNA、RNA和蛋白質。如果對一個很少的標本要進行這三種物質分析,Trizol試劑是**佳的選擇。
【材料】
Trizol試劑,氯仿,異丙醇,75%的乙醇,DEPC處理水,PBS緩沖液。
【操作程序】
(1) 收獲細胞5×106個,方法同DNA提取。
(2) 在含有細胞團的Eppendorf管中加入1m1Trizol試劑,振蕩混勻,室溫放置5分鐘(為了獲得好的結果,可在15℃水浴中進行)。
(3) 加氯仿0.2ml,振蕩混勻,室溫放置2分鐘。
(4) 4℃,12 000g離心15分鐘,小心吸取上層水相,移人另一Eppendorf管中(不可觸及中間層的蛋白質界面)。
(5) 加入等體積的異丙醇,置室溫10分鐘。
(6) 4℃,12 000g離心15分鐘,棄上清。
(7) 冰浴冷的75%乙醇漂洗沉淀;4℃,12 000g離心5分鐘,棄上清。
(8) 真空干燥,去除樣品中的乙醇,用適量的DEPC處理水溶解沉淀,測定OD260、280,計算RNA的含量和純度,同時取大約150ng總RNA電泳檢測完整性。
【注意事項】
此兩種方法是目前常用且比較簡單的RNA提取方法,在提取的時候,**好是先計算細胞的數目,決定變性液或Trizol試劑的用量,如果細胞數目過多,常出現在RNA中有蛋白質和DNA的污染。不論采用何種方法,都很難保證沒有DNA污染,在做RT-PCR時將出現非特異性擴增和雜帶,所以推薦在RNA提取后,用DNAseI(無RNase活性)消化DNA,獲得高純度的RNA。如果從轉染有質粒或病毒的細胞,則必須用DNAseI消化。
3.蛋白質提取 蛋白質是基因的**終產物,是功能的執(zhí)行者。在研究中需從細胞中提取蛋白質進行酶的活性分析和產物鑒定。下面介紹同和種常用的方法。
方法一:SDS-超聲
此種方法利用SDS裂解細胞,再用超聲的方法斷裂DNA,所獲得蛋白質溶液可以用于SDS-PAGE分析和Western blot。
【材料】
1×SDS加樣緩沖液:50mmol/L Tris-Cl (pH6.8),100mmol/L DTT(二硫蘇糖醇),2%SDS,0.1%溴酚蘭,10%甘油。PBS緩沖液。
【操作程序】
(1) 棄細胞培養(yǎng)基,用PBS洗滌細胞2次。
(2) 用橡膠刮下細胞(不要用胰蛋白酶消化),加入適量的PBS緩沖液,離心收獲細胞。
(3) 在含有細胞團的離心管中加入150μl 1×SDS 加樣緩沖液,此時溶液比較粘稠,是由于大量基因組的DNA的釋放。
(4) 將上述溶液煮沸5~10分鐘。
(5) 用超聲波打碎染色體DNA,直到溶液不粘稠為止。
(6) 室溫下10 000g離心10分鐘,取上清到另一個離心管中。
(7) 測定蛋白質濃度。所得溶液可以用SDS-PAGE分析和Western blot。
方法二:Trizol法
Trizol試劑除了可以分離DNA和RNA外,還可用于蛋白質的提取。
【材料】
含0.3mol/L鹽酸胍的95%乙醇。
【操作程序】
(1) 在Trizol法DNA提取過程中⑦的上清(酚和乙醇相)中,加入1.5ml的異丙醇,室溫10分鐘。
(2) 4℃,12 000g離心10分鐘。
(3) 棄上清,用含0.3mol/L鹽酸胍的95%乙醇溶液洗滌沉淀3次。在每次洗滌過程中,沉淀在溶液中保持20分鐘。
(4) 4℃,10 000g離心5分鐘。
(5) 用2ml的無水乙醇洗滌沉淀1次,4℃,10 000g離心5分鐘。
(6) 吸去上清,真空干燥,用1%SDS溶解蛋白質沉淀。
(7) 4℃,12,000g離心10分鐘除去非需物質,轉移上清(即含蛋白質的溶液)。
【材料】
細胞裂解液:Tris-Cl (PH 8.0) 50mmol/L,NaCl 150mmol/L, 0.02%Na3N,PMSF 100μg/ml,1% Triton X-100,PBS緩沖液。#p#分頁標題#e#
【操作程序】
(1) 離心收獲對數期生長細胞,用PBS洗滌細胞2次。
(2) 向細胞沉淀加入800μl細胞裂解液,4℃放置30分鐘(為了進一步破碎細胞,可用超聲破碎機粉碎)。
(3) 4℃、12 000g離心30分鐘,吸上清,即為蛋白質液。
【注意事項】
此種方法用了蛋白酶抑制劑PMSF(苯甲酰磺酰氟),沒有應用蛋白質變性劑,所以此法所得的蛋白質液可以用于酶的活性分析,但其和Na3N有劇毒,實驗操作要小心,實驗過程中要帶手套謹慎操作。
二、基因導入
當一個基因被克隆后,研究者總是希望將其轉染到真核細胞中進行研究其功能、表達調控、分離蛋白質產物等。所有這些目的,都必須具有將DNA有效地導入細胞的能力。目前基因導入技術已經廣泛地用于基因結構與功能分析,基因表達與調控、基因治療與轉基因動物研究,已經成為分子生物學研究的一種常用的手段。
這里介紹幾種常用的細胞基因轉染的方法。
方法一:脂質體轉染
轉染用試劑脂質體Lipofeetin和Lipofectamine是由Invitrogen 公司推出的**代產品和第二代產品。Lipofectamine與Lipofectln相比,毒性更小,轉染效率更高,特別適合于一些轉染的細胞,如HeLa、COS-7、NIH3T3、PC12D等。脂質體是一種特制的陽離子脂質試劑,其與靶DNA的磷酸骨架結合而成的復合物,具有能輕易通過細胞膜從而完成轉染過程的特性。可轉染DNA、RNA和寡核苷酸**各種細胞,并將DNA導入植物原生質體。目前在研究基因功能方面,出現一個新的技術是RNA干擾試驗(RNA interference),在操作過程中,是將21 ~ 25nt寡核糖核酸雙鏈分子轉染細胞,從而穩(wěn)定、特異性去除某一個基因的表達,然后從細胞的表現中推測未知基因的功能。所用的轉染試劑就是Lipofectamine。脂質體適用于各種類型的貼壁生長和懸浮培養(yǎng)的細胞,其介導的轉染效率是磷酸鈣沉淀法德5 ~100倍。
1.貼壁細胞轉染方法
【材料】
脂質體,不完全培養(yǎng)基(RPMI 1640),完全培養(yǎng)基(含15%胎牛血清)。
【操作程序】
(1) 質粒的制備:由于用于轉染的質粒量比較大,所需的純度也較高,而且質粒的構型**好是共價閉環(huán)的。所以推薦采用質粒DNA大量制備,而且在操作過程中要輕柔。提取的質粒需進行純化,如用氯化銫密度離心法、聚乙二醇沉淀法。目前市上所售的一些質粒提取和純化的試劑盒可以達到基因轉染的要求,并且操作比較簡單、快捷。在質粒純化的**后一步質粒沉淀時,應是無菌操作,70%的乙醇漂洗,無菌超凈臺內吹干,溶于無菌水中。
(2) 收獲細胞:將(1 ~ 2)×105個細胞重懸于2ml完全培養(yǎng)基中,轉種于35mm培養(yǎng)皿中或6孔培養(yǎng)板中。
(3) 37℃,5%CO2培養(yǎng)箱培養(yǎng)18 ~ 24小時,使細胞達50%~80%的融合。
(4) 在Eppendorf管中,制備下列溶液:A溶液:將2~20μg質粒DNA溶于100μl RPMI 1640培養(yǎng)基中。B溶液:Lipofectin[Lipofectin(μl):DNA(μg)約為2.5:1 ]稀釋于 RPMI l640培養(yǎng)基中,**終體積100μl。
(5) 合并A溶液和B溶液,輕輕混勻,置于室溫15分鐘。
(6) 棄去培養(yǎng)皿或培養(yǎng)板中的細胞培養(yǎng)液,并用無血清培養(yǎng)基洗滌細胞一次。
(7) 加0.8ml無血清培養(yǎng)基**Lipofectin-DNA混和物中,混勻后,小心滴在細胞上,輕輕混勻。
(8) 37℃,5%CO2培養(yǎng)箱培養(yǎng)5~24小時(這個時間要根據細胞耐受無血清培養(yǎng)時間而定)。
(9) 棄去轉染液,加入2ml完全培養(yǎng)基,繼續(xù)培養(yǎng).
(10) 轉染后48~72小時,測定細胞瞬時表達情況,如用于穩(wěn)定表達,可于轉染48小時后更換選擇培養(yǎng)基進行篩選。
2.懸浮細胞一過性轉染方法
【材料】
同貼細胞轉染方法。
【操作程序】
(1) 質粒的制備同上。
(2) 收獲細胞,用無血清培養(yǎng)基洗滌細胞1次。
(3) 將(2 ~ 3)×106個細胞重懸于0.8ml無血清培養(yǎng)基中,接種于6孔板或35mm培養(yǎng)皿中。
(4) 參照貼壁細胞轉染方法(4)、(5)進行制備Lipofectin-DNA混和物。
(5) 將制備的Lipofectin-DNA混和物加入細胞懸液中,輕輕混勻,置37℃,5%CO2培養(yǎng)箱培養(yǎng)5 ~ 24小時。
(6) 加4ml完全培養(yǎng)基繼續(xù)培養(yǎng)。
(7) 轉染48 ~ 72小時后,室溫200g,離心5分鐘收獲細胞。測定細胞瞬時表達情況。
Lipofectamine與Lipofectin轉染的方法基本相同,不同公司的產品有稍許不同,請參照其產品說明書進行,表13-1列出不同直徑的培養(yǎng)皿的轉染過程所需的試劑量。
表13-1 不同培養(yǎng)皿所需試劑量
|
培養(yǎng)皿直徑 |
脂質混合物 |
Lipofectamine或Lipofectin(μl) |
DNA |
無血清培養(yǎng)基 |
|
35 |
100 |
2-25 |
1-2 |
0.8 |
方法二:磷酸鈣沉淀法
此法是研究基因轉染**先采用的技術,操作簡單,不需昂貴的轉染試劑,**今仍有研究者應用此技術。其機制可能是DNA與磷酸鈣形成沉淀物,使之粘附到培養(yǎng)的哺乳動物細胞表,被細胞內吞。但此法存在著不足的地方就是轉染效率比較低,有些細胞不能用此種方法進行轉染。
l.貼壁細胞轉染方法
【材料】
(1) 2mol/l氯化鈣,0.22μm濾膜過濾除菌,-20℃保存。
(2) 2×HBS緩沖液:280mmol/L NaCI,10mmol/L KCl,1.5 mmol/L Na2HPO4,12mmol/L葡萄糖,50mmol/L HEPES。
將1.6g NaCI,0.074g KCl,0.027g Na2HP04·2H20,0.2g葡萄糖,1g HEPES溶于90ml水中,用0.5mol/L NaOH調節(jié)pH為7.0,然后用雙蒸水定容**l00ml,0.22μm濾膜過濾除菌,分裝,-20℃保存。
(3) 含15%甘油的1×HBS緩沖液。
(4) PBS緩沖液(pH7.4)。
【操作程序】
(1) 在轉染前24小時,將對數生長期細胞用胰酶消化,將1×106個細胞接種于60mm的培養(yǎng)皿中,置37℃、5%CO2培養(yǎng)箱中繼續(xù)培養(yǎng)。
(2) 在轉染前2小時,按表13-2配制DNA-磷酸鈣共沉淀物
表13-2 DNA-磷酸鈣共沉淀物的制備
|
試劑 |
60mm培養(yǎng)皿 |
100mm培養(yǎng)皿 |
試劑 |
60mm培養(yǎng)皿 |
100mm培養(yǎng)皿 |
|
DNA(μg) |
6~12 |
10~20 |
2mol/L氯化鈣(μl) |
37 |
62 |
將質粒DNA于0.263ml無菌水中,加入0.37μl 2mol/L氯化鈣溶液,混勻后,緩慢加入2×HBS緩沖液,并不斷輕輕搖振,緩沖液在30秒內加完。將所配制的混和物在室溫靜置30分鐘。
(3) 棄去細胞培養(yǎng)基,用無血清培養(yǎng)基或PBS緩沖液洗滌一次,將DNA-磷酸鈣沉淀物加入培養(yǎng)皿中,在室溫下靜置20~30分鐘,然后再加入5ml培養(yǎng)基,37℃、5%CO2培養(yǎng)箱中繼續(xù)培養(yǎng)。
(4) 培養(yǎng)24~48小時,可進行瞬時表達檢測,或用適當的選擇性培養(yǎng)基進行穩(wěn)定轉化克隆的篩選。
2.懸浮培養(yǎng)細胞轉染方法
(1) 用離心法收獲1×107個細胞,棄細胞培養(yǎng)基,用PBS緩沖液洗滌一次細胞沉淀,再一次離心收獲細胞。
(2) 制備DNA-磷酸鈣沉淀物,方法同上。
(3) 用0.5ml DNA-磷酸鈣沉淀物重懸細胞沉淀,置室溫10~20分鐘。在細胞懸液中加入5ml完全培養(yǎng)基,置37℃、5%CO2培養(yǎng)箱中繼續(xù)培養(yǎng)。
(4) 培養(yǎng)16~24小時,收集細胞,棄轉染液,換以完全培養(yǎng)液繼續(xù)培養(yǎng)。
(5) 37℃、5%CO2培養(yǎng)箱中培養(yǎng)48小時后,進行表達檢測或者克隆化篩選。
【注意事項】
(1) 在制備DNA-磷酸鈣沉淀物的靜置過程中,約5分鐘出現輕度混濁,濁度越來越深,大約在20~30分鐘時,在顯微鏡下觀察出現小顆粒。
(2) 顆粒的大小與轉染的效率密切相關,其判定方法是將試管對著光線觀察,見溶液呈混濁狀態(tài),略帶白色,但肉眼又看不見顆粒,在高倍顯微鏡下則可見均勻細小的顆粒,此時的顆粒為比較理想的狀態(tài)。如果用肉眼即能看到顆粒,則說明所形成的顆粒太大;如果20分鐘后溶液仍然透明,則說明無顆粒形成或形成的顆粒太小。
(3) 在加入2×HBS時,一定要不斷的振搖,否則會形成大塊狀的顆粒。
(4) 在配制2×HBS緩沖液時,一定要注意溶液的pH,其可明顯地影響沉淀顆粒的形成。偏酸則不能形成顆粒,偏堿則形成的顆粒太大。這兩種情況均可導致轉染失敗。
(5) 在實際操作中,有的研究者為了增加轉染的效率,在轉染的(3)步驟后2~4小時進行甘油休克。在實驗前需要進行預實驗,檢測該細胞對甘油的敏感性。方法是:收集對數生長期細胞1×107個,棄培養(yǎng)基,加入0.5ml含15%甘油的1×HBS緩沖液。37℃溫育,分別在顯微鏡下觀察1、2、3分鐘時的細胞變化,如果在溫育過程中,發(fā)現細胞變圓并死去,則轉染后勿進行甘油休克。如果細胞形態(tài)未發(fā)生明顯變化,才可進行甘油休克。方法同細胞對甘油的敏感性檢測。甘油休克后,棄15%甘油的l×HBS緩沖液,用PBS洗一次,加入完全培養(yǎng)基繼續(xù)培養(yǎng)。
(6) 另個增強轉染效率的方法是用氯哇處理細胞。氯喹對細胞具有毒性作用,一般在實驗前需進行預實驗決定合適的濃度。但對于大多數細胞來說,用濃度為l00μmol/L的氯喹處理或取得良好的效果。可在DNA-磷酸鈣沉淀物加入細胞之前或之后進行,但需在甘油休克之前。處理后用PBS液清洗。在處理的過程中細胞會出現泡狀變化,這是正常現象。#p#分頁標題#e#
方法三:電穿孔轉染技術
電穿孔是指在高壓電脈沖的作用下使細胞膜上出現微小的孔,導致不同細胞之間的細胞膜發(fā)生融合。在后來的研究中發(fā)現,對細胞進行電擊可以促使細胞通過微孔吸收外界環(huán)境中的DNA分子,并進入細胞核內部。
【操作程序】
(1) 在10%胎牛血清DMEM的培養(yǎng)基中生長**50%~80%融合,收集細胞(0.5~1.0)×107/ml,并用冰預冷的電穿孔緩沖液冼滌細胞2次。常用的電擊緩沖液有:PBS緩沖液(pH7.4),磷酸鹽蔗糖緩沖液(272nmol/L,7mmol/L磷酸鈉pH7.4,1mmol/L MgCl2),Hamm’s Fl2培養(yǎng)基(不含小牛血清和抗生素)。
(2) 取0.8ml細胞懸液放人0.4cm的基因脈沖小池(gene pulser cuvette)中,取3~40μg質粒加入細胞懸液中,充分混勻,冰浴10分鐘。
(3) 將基因脈沖小池放在電脈沖儀的正負極之間,電擊1次,電擊條件依據電擊緩沖液有所不同(表13-3)。
表13-3 所用的緩沖液及所需的電壓
|
緩沖液 |
電容(μF) |
電壓(V) |
緩沖液 |
電容(μF) |
電壓(V) |
|
PBS緩沖液 |
25 |
100~1600 |
Hamm’s F12培養(yǎng)基 |
960 |
250~450 |
(4) 電擊后將小池冰浴10分鐘。
(5) 從小池中吸出細胞,并用適量的培養(yǎng)墓稀釋細胞,37℃、5%CO2培養(yǎng)箱中培養(yǎng)48小時后,進行表達檢測或者克隆化篩選。
【注意事項】
(1) 電擊的**大電壓和電擊持續(xù)時間是影響轉染效率的2個主要因素。電擊電壓太小和/或持續(xù)時間太短,則轉染效率太低;如果電擊成壓太大和/或電擊時間太長,細胞將不能存活,所以用此種方法轉染為了取得好的轉染效率,須進行反復實驗摸索出**佳的電擊電壓和電擊持續(xù)時間。
(2) 細胞處于有絲分裂期時比較易感染外源DNA,所以在進行轉染時,細胞**好處于對數生長期。
(3) 質粒DNA的狀態(tài)對轉染的影響。要使外源DNA整合到細胞染色體上,**好用線性DNA(因為線性DNA有較高的重組幾率)。瞬時表達用環(huán)狀的DNA即可。
(4) 質粒DNA的濃度在2~40μg/ml范圍內時,隨著DNA的濃度的增加,轉染效率隨著增加。作為穩(wěn)定表達時,DNA的濃度為2~10μg/ml,而某些瞬時表達系統(tǒng)則需20~40μg/ml。
基因導入的方法還有DEAE-葡聚糖轉染技術、基因顯微注射和納米級分子級顆粒的非脂質體型轉染試劑等方法。DEAE-葡聚糖轉染的方法一般用于基因瞬時表達,它不易形成穩(wěn)定轉染的細胞系。基因顯微注射需要特殊的儀器和設備,操作比較復雜,但這種方法是建立轉基因動物模型以研究外源基因在整體動物中表達調控規(guī)律的**好方法。同時可以改變動物的基因型,更符合人類的需要,使轉基因動物產生人類所需的生物活性物質。納米級分子級顆粒的非脂質體型轉染試劑是一種不含脂類分子、內毒素及任何動物來源的成分,當它與DNA混合時,形成納米大小的轉染復合物,這種技術由于不含脂類分子,所以特別適用于研究脂類分子的實驗、信號轉導研究及制藥公司藥物篩選。
篩選細胞克隆:
基因轉染人細胞后,有些細胞克隆所轉染的基因是高表達,而有些克隆是低表達。有些研究需要對穩(wěn)定轉染的細胞克降進行篩選。一般需要2個過程,一是藥物篩選,二是克隆細胞株的轉移和擴增。
1.藥物篩選 在基因導人過程中只有一小部分細胞獲得了外源性的DNA,穩(wěn)定整合到細胞的基因組DNA中,并且表達產物。為了有效、方便地鑒定出這些細胞來,一般在載體上具有一個能在這些細胞中產生可選擇性變化的顯性遺傳標志。目前常用的這種標志有胸腺核苷激酶基因(tk)、二氫葉酸還原酶基因(dhfr)、氯霉素乙酰轉移酶基因(cat)和新霉素磷酸轉移酶基因(neo)。表13- 4列出了這些標記所適用的細胞和所用的篩選藥物。
表13-4 標記基因所適用的細胞和所用的篩選藥物
|
標志基因 |
篩選藥物 |
所適用的細胞 |
標志基因 |
篩選藥物 |
所適用的細胞 |
|
胸腺核苷激酶基因 |
HAT |
tkˉ細胞 |
氯霉素乙酰轉移酶基因 |
|
所有真核細胞 |
氯霉素乙酰轉移酶基因檢測系統(tǒng)不需加入篩選藥物,但需在制備的細胞提取物中加入檢測試劑乙酰輔酶A和14C標記的氯酶素進行孵育,薄層層析后通過放射自顯影測定產物。所以此法常用來分析啟動子活性、表達產物的量等,一般用在瞬時表達研究。其他3種用來研究穩(wěn)定基因表達,**常用的是新霉素磷酸轉移酶基因標記系統(tǒng)。下面介紹的是G418篩選的方法。
【操作程序】
(1) 取l00mg G418溶于2ml去離子水中(濃度為50mg/m1),0.22μm微孔濾器過濾除菌,-20'C保存。
(2) 轉染后經過48~72小時,待細胞生長接近融合時按1︰4傳代。
(3) 繼續(xù)培養(yǎng)**細胞達50%~70%融合。
(4) 棄去培養(yǎng)液,更換含有800μg/ml的G418培養(yǎng)液進行篩選(其濃度可依據預實驗來確定,不同的細胞對G418有不同的適合濃度),與此同時用未轉染的細胞作對照進行。
(5) 當大部分細胞死亡時(約3~5天后,在鏡下觀察,細胞不再貼壁,漂浮起來,細胞變圓) ,再換一次液,G418濃度可降**150~250μg/ml,以維持篩選作用。
(6) 約10~20天后,可見有抗性克隆形成,待其逐漸長大后,將其轉移和擴增。
2.克隆細胞株的轉移和擴增
將所形成的克隆進行擴增培養(yǎng)的方法常用的有兩種方法。
方法一:胰酶–濾紙粘附
【操作程序】
(1) 將所形成的克隆在顯微鏡下準確標記位置。
(2) 在超凈臺內,吸去培養(yǎng)基,并用無血清的培養(yǎng)基洗滌兩次。
(3) 用無菌鑷夾取一塊約5mm方形滅菌3MM濾紙,用0.25%胰酶浸濕,置于克隆所標記的位置處,5~20秒。
(4) 將24孔培養(yǎng)板每孔加入含有250μg/ml G418的選擇性培養(yǎng)基2ml,用鑷子取出粘附有克隆細胞的濾紙塊,置于24孔培養(yǎng)基中,涮洗數次,以使濾紙上粘附的細胞脫下。
(5) 將移有克隆細胞的24孔培養(yǎng)板置于37℃、5%CO2培養(yǎng)箱中繼續(xù)培養(yǎng)。
(6) 待細胞長滿后,胰酶消化后,進一步擴大培養(yǎng)。
方法二:胰酶–Tip頭吸取
(1) 細胞克隆位置的確定和處理同上(1)和(2)。
(2) 用微量移液29(帶無菌Tip頭)吸取2~5μg 0.25%胰酶滴在細胞克隆位置上,并反復吹打數次,吸取液體。
(3) 將含有細胞的胰酶液體含有加入250μg/ml G418的選擇性培養(yǎng)基2ml 24孔板中。37℃、5%CO2培養(yǎng)箱中繼續(xù)培養(yǎng)。
(4) 待細胞長滿后,胰酶消化后,作進一步擴大培養(yǎng)、鑒定。
上述列出了一些常用的基因導人方法,其結果是否有效地轉染,除了用藥物抗性篩選標志外,還需對其產物進行檢測,進一步鑒定,所用的方法就是應用細胞的分離提取研究,提取RNA進行RT-PCR檢測和Northern blot檢測,提取蛋白質進行Western blot檢測。另外還有下面將要介紹的原位檢測。
三、 原位檢測研究
對有些很難大量培養(yǎng)細胞的基因和其產物檢驗和鑒定,很難用提取蛋白質和核酸的方法進行,需要用靈敏度比較高的原位檢測的方法。根據檢測的對象可以分為兩類,一是蛋白質檢測,應用原理是抗體與抗原的特異結合,它不但反應蛋白質的表達量,還可以反應蛋白質所處位置;二是核酸檢測,應用原理是用標記好的核酸探針與細胞巾的核酸分子進行雜交反應。
1.蛋白質檢測 此種方法根據二抗的類型,可以分為化學檢測和熒光檢測,由于熒光檢測的靈敏度比較高,而且也比較方便、簡便,所以現較多采用。下面介紹的就是免疫熒光檢測。
【操作程序】
(1) 取干凈蓋玻片,放人75%乙醇浸泡1小時,取出玻片在酒精燈上烤干,用DMEM培養(yǎng)基漂洗數次,放人細胞培養(yǎng)皿中,將傳代的細胞傳人該皿。
(2) 待蓋玻片上長滿細胞后,取出玻片用PBS緩沖液洗2~3次,,空氣干燥(一定要干燥)。
(3) 將蓋玻片浸入-20℃預冷的丙酮內,于-20℃固定30分鐘,PBS緩沖液洗3次,空氣晾干(必須要于燥)。
(4) 在parofilm膜上滴加**抗體,將蓋玻片有細胞的一面向下蓋在抗體上,密封于37℃濕盒中孵育30分鐘~1小時。
(5) PBS液洗3~6次,空氣中晾干。
(6) 加熒光素標記的第二抗體在parafilm膜上,將蓋玻片有細胞的一面向下蓋在抗體上,放入濕盒中,于37℃孵育30分鐘~1小時。
(7) PBS液洗3~6次,空氣中晾干。
(8) 用60%的甘油將蓋玻片封在載玻上在熒光顯微鏡下觀察。
【注意事項】
(1) 細胞的處理可以在收獲后涂片在載玻片上,進行丙酮固定;加抗體于載玻片上,可用蓋玻片蓋住抗體進行反應。
(2) 所加抗體的量要根據預實驗來確定,第二抗體在用時一般需進行稀釋,—般以1︰100~1000為宜。
(3) 免疫熒光檢測靈敏度高、簡便,但有時有較多的非特異性熒光顆粒,所以—定要做陰性對照,可以將**抗體換為其他的無關抗體。
(4) 由于熒光素在自然光下易于褪色,所以實驗結果應立即觀察,若要保存可將玻片包在黑紙內,置4℃冰箱可保存一周。
(5) 這種熒光檢測的方法,有時熒光素標記在**抗體上,則可不用第二抗體,成之為直接免疫熒光檢測,此時可以使實驗的時間縮短,又可以明顯降低非特異反應。上述介紹的是間接熒光免疫檢測。
2.核酸檢測 這種核酸檢測亦稱之為細胞核酸原位雜交,其基本原理是利用核酸分子的堿基序列配對的互補性(A:T,A:U,G:C),將已知的有標記的外源核酸分子(探針)與細胞標體內的RNA或DNA進行雜交,經過放射自顯影或酶的催化反應顯示其在細胞的位置。細胞原位雜交技術近年束發(fā)展非常迅速,尤其在發(fā)育生物學、遺傳學、病毒學及腫瘤學等*域中應用廣泛。探針的標記技術和檢測技術也不斷地更新。由放射性探針到目前許多非放射性探針,如生物素蛋白–AP(堿性磷酸酶)、地高辛–AP和地高辛–HRP(辣根過氧化物酶)等檢測系統(tǒng)。根據探針的標記物是否能夠直接被檢測,原位雜交技術可以分為直接法和間接法兩種。為了提高檢測技術的敏感度,將PCR技術與原位雜交結合起來,大大提高了檢測效率。下面主要介紹培養(yǎng)細胞的原位雜交地高辛–堿性磷酸酶檢測技術。#p#分頁標題#e#
【操作程序】
(1) 細胞片的制備:
1) 將蓋玻片上生長的細胞用PBS液洗滌3次,空氣中干燥。亦可取細胞培養(yǎng)瓶中胰酶消化的細胞制成單細胞懸液,涂片到載玻片上;對于懸浮培養(yǎng)的細胞將細胞涂片**載玻片上,此時所用的載玻片要預先涂有多聚賴氨酸。
2) 用1︰1的甲醇:丙酮或4%的多聚甲醛固定10~30分鐘。
3) 0.2moL HCl室溫作用10分鐘,0.1%~0.3%TritonX-100作用15分鐘。
4) 含0.2%的甘氨酸的PBS液于室溫孵育15分鐘。
5) 1μg/ml蛋白酶K(0.1mo/L Tris-Cl,50mmol/L EDTA,pH8.0 配制) 37℃濕盒中消化10~30分鐘。
6) 含0.2%甘氨酸的PBS液于室溫孵育15分鐘。
7) PBS-5mmol/L MgCl2洗2次,每次10分鐘。
8) 4%的多聚甲醛固定15分鐘。
9) PBS-5mmol/L MgCl2洗2次,每次10分鐘。
10) 逐級酒精脫水(70%、80%、90%、95%、100%各5分鐘),空氣中干燥。
(2) 預雜交和雜交:
1) 將玻片浸入2×SSC中15分鐘。
2) 再用50%去離子甲酰胺(4×SSC與甲酰胺等體積配制)37℃孵育15分鐘。
3) 加預雜交液20μl/片42℃孵育30分鐘~1小時。試劑盒中有此試劑,亦可自行配制,50%去離子甲酰胺,5×SSC,5×Denhardt’s液,2%SDS,100μg/ml變性的鮭魚精DNA。
4) 去除預雜交液,每片加20μl雜交液(含0.2 ~520μg/ml探針),用硅化過的蓋玻片覆蓋,石蠟封住蓋玻片的四周,42℃濕盒中孵育12~18小時。
5) 去除石蠟,小心移去蓋玻片(可以在2×SSC浸泡,讓其自行脫落),用2×SSC洗2分鐘。
6) 在含20μg/ml RNase的2×SSC溶液中于37℃浸泡30分鐘 (此步針對RNA探針,對于DNA探針省略)。
7) 2×SSC 42℃洗滌,10分鐘,2次。
8) 0.1×SSC 42℃洗滌2次,每次30分鐘,
(3) 雜交檢測:
在進行檢測前需配制下列緩沖液。緩沖液I:順丁烯二酸(馬來酸)0.1mol/L,NaCl0.15mol/L,用NaOH將pH調**7.5(20 ℃),高壓滅菌。
緩沖液Ⅱ:將阻斷劑溶于緩沖液I中**終深度為10%(W/V),加熱溶解,高壓消毒。用水10倍稀釋即成緩沖液Ⅱ。
緩沖液Ⅲ(pH9.5,20℃):Tris-Cl 100mmol/L,NaCl l00mmol/L,MgCl2 50mmol/L。
TE緩沖液:Tris-Cl 100mmol/L,EDTA 1mmol/L。
顯色液:取45μl NBT(硝基四氮唑藍),35μl X-phosphate-solution(5溴-4氯-3吲哚磷酸,BCIP),加10ml緩沖液Ⅲ混和而成(現配現用)。
【操作程序】
1) 將玻片用緩沖液I洗滌,室溫,2分鐘。
2) 將玻片用緩沖液Ⅱ洗滌,室溫,30分鐘,不斷振搖。
3) 用緩沖液Ⅱ將地高辛抗體–堿性磷酸酶1︰5000稀釋,將玻片浸入此溶液中1~2小時。
4) 緩沖液I洗滌,室溫,15分鐘,2次。
5) 緩沖液Ⅲ洗滌,室溫,3分鐘。
6) 將玻片浸入顯色液中,室溫,避光顯色,16小時(在顯色過程中,不要晃動液體)。
7) TE緩沖液洗滌,室溫,2分鐘。
8) 用親水性封片劑封片,顯微鏡下觀察。陽性反應呈深藍色。
綜上所述,本節(jié)詳細描述了細胞培養(yǎng)過程小的分子生物學研究操作步驟。如前所述,任何基因的功能,都需要通過細胞內的過程加以研究和檢測,因而細胞培養(yǎng)技術是分子生物學的不可缺少的一個重要部分。
第二節(jié) 細胞培養(yǎng)在生物工程中的應用
生物工程是指用生物體或其組成成分自**適條件下生產有益產物或進行有效過程的技術它所包含的內容有主要有基因工程、酶工程、發(fā)酵工程、細胞工程、生化工程等。近年來把細胞的大量培養(yǎng)亦列為細胞工程的內容。在這些內容中與動物細胞培養(yǎng)相關的主要是細胞工程。
細胞工程是生物工程的一個重要方面。總的來說,它是應用細胞生物學和分子生物學的理論和方法,按照人們的設計藍圖,進行在細胞水平上的遺傳操作及進行大規(guī)模的細胞和組織培養(yǎng)。當前細胞工程所涉及的主要技術*域有細胞培養(yǎng)、細胞融合、細胞拆合、染色體操作及基因轉移等方面。通過細胞工程可以生產有用的生物產品或培養(yǎng)有價值的細胞株,并可以產生新的物種或品系。
細胞工程已經滲透到人類生活的許多*域,取得了許多具有開發(fā)性的研究成果,有的已在生產中推廣,收到了明顯的經濟和社會效益。隨細胞工程技術研究的不斷深入,它的前景和產生的影響將會日益地顯示出來。
根據設計要求,按照需要改造遺傳物質的不同操作層次,可將細胞工程學分為染色體工程、染色體組工程、細胞質工程和細胞融合工程等幾個方面。
1.染色體工程 染色體工程是按人們需要來添加、削減或者替換同源或異源染色體或其中的某一部分的方法技術。可分為動物染色體工程和植物染色體工程兩種。動物染色體工程主要采用對細胞進行微操作的方法(如微細胞轉移方法等),來達到轉移基因的目的。植物細胞工程目前主要是利用傳統(tǒng)的雜交回交等方法來達到添加、消除或置換染色體的目的。
2.染色體組工程 染色體組工程是按照人們的設計,削減或添加同種或異種染色體組,以改變生物遺傳物質的技術和方法。經過這種遺傳物質改造的物種,會符合人們的需要,如四倍體水稻的稻粒比二倍體的明顯大,蛋白質的含量可提高5%~50%。但這項技術一般應用在植物上。#p#分頁標題#e#
3.細胞質工程 又稱細胞拆臺工程,按照人們的沒想,運用物理或化學方法將細胞質與細胞核(或細胞器,如線粒體)分開,再進行不同細胞間核質的重新組合,重建成新細胞。可用于研究細胞核與細胞質關系的基礎研究和育種工作。克隆羊“多莉”就是一個很好的例子,此項技術又稱核移植技術。
4.細胞融合工程 是用自然或人工的方法使兩個或幾個不同細胞融合為一個細胞的過程。可用于產生新的物種或品系(植物上用得多,動物上用得少)及產生單克隆抗體等。其中單克隆抗體技術,利用克隆化的雜交瘤細胞分泌高度純一的單克隆抗體,具有很高的實用價值,在診斷和治療病癥方面有著廣泛的應用前途(此項內容在細胞培養(yǎng)生物制品的應用中有介紹)。另外細胞融合也應用在基因功能和尋找致病基因的研究中,如小鼠骨髓瘤細胞與人淋巴細胞融合后的雜種細胞總是保留著小鼠完整染色體組型,而僅有一條**數條人染色體。根據染色體分析技術對雜種細胞及親本瘤細胞生物功能(如酶譜等)分析,說明是由于人染色體整合到瘤細胞染色體中所引起的。同時細胞融合技術也可用于分析癌基因在染色體中所處的位置,目前已知腫瘤基因可誘發(fā)腫瘤,采用細胞融合及染色體分子技術測定細胞致癌染色體,即可判斷致癌基因在染色體上所處位置。因此,細胞融合技術對研究染色體結構,闡明生物變異及腫瘤發(fā)生與發(fā)展機制有一定指導意義。
細胞培養(yǎng)除了在細胞工程應用**多之外,在酶工程中亦有應用,即將細胞限制在特定的空間位置,與酶同樣起到生物催化劑的作用。此項技術稱之為固定化細胞技術。這種技術由細菌已經擴展到動物細胞,甚**到細胞器,如線粒體、微粒體等。
大規(guī)模的細胞培養(yǎng)技術是細胞工程的一個內容,指在人工條件下,搞密度大量培養(yǎng)有用動物細胞,生產有應用價值的細胞產品的技術,如疫苗(口蹄疫苗、狂犬病毒疫苗、脊髓灰質炎疫苗、牛白血病病毒疫苗、乙型肝炎病毒疫苗、皰疹病壽I型及Ⅱ型疫苗、巨細胞病毒疫苗等)、蛋白質因子(凝血因子Ⅷ和Ⅸ、促紅細胞生成素、生長激素、IL-2、神經生長因子等)、免疫調節(jié)劑(α、β、γ干擾素)及單克隆抗體等;也是生物工業(yè)中大量增殖新型有用細胞不可缺少的技術,一些培養(yǎng)細胞可用于治療,如劉書欽等建立和應用大規(guī)模培養(yǎng)系統(tǒng),誘導獲得了移植人的CTL(細胞毒性T淋巴細胞),具有抗人肺鱗狀癌腫SQ-5能力,保存3個月以后仍保持很高殺傷活性,達95%以上。在本節(jié)中主要介紹細胞的大規(guī)模培養(yǎng)。
當人們認識到利用細胞培養(yǎng)可以產生激素、疫苗、單克隆抗體時,而且對它們的需要量急劇增加,細胞的大規(guī)模培養(yǎng)應運而生了。開始是單靠增加容器的體積和數量來解決細胞產量的問題,隨著對動物細胞反應動力學的認識和培養(yǎng)技術的改進,使得動物細胞工業(yè)化培養(yǎng)成為現實并逐漸成熟,目前在生產規(guī)模上已經達到10 000L。
一、大規(guī)模細胞培養(yǎng)的體系參數
動物細胞的大量培養(yǎng)與小規(guī)模培養(yǎng)(培養(yǎng)容量小于2L)相比,條件更嚴格,控制難度更大,但培養(yǎng)的條件有些與小量培養(yǎng)是一致的。常用的參數有:選用的培養(yǎng)基是無血清培養(yǎng)基,對于懸浮培養(yǎng)的細胞,為使其細胞不致凝集、成團或沉淀,在配制培養(yǎng)基的基礎鹽溶液中不加Ca2+和Mg2+。加入一些非營養(yǎng)性的培養(yǎng)液補充物,如0.11%的羥甲基纖維鈉,它可以減輕在培養(yǎng)過程中由于機器的攪拌造成對細胞的剪切損傷。0.11%的pluronic F–68,它可以減少在通氣和攪拌過程中所產生的氣泡。一些特殊的細胞需加入一些營養(yǎng)因子,如轉鐵蛋白、胰島素、亞硒酸鹽等。細胞培養(yǎng)溫度足37℃,在細胞接種前培養(yǎng)液需預溫**37℃。細胞的接種密度為(5~20)×104個/ml。攪拌速率依據培養(yǎng)容器和細胞的培養(yǎng)方式決定,一般對懸浮細胞可用100~500rpm,而微載體系統(tǒng)20~100rpm。pH一般控制在7.4,但有些細胞偏好在7.0,如雜交瘤,但不能低于6.8,否則將抑制細胞的生長。pH的穩(wěn)定維持一般是依靠CO2-NaHCO3系統(tǒng),采用通入5%CO2無菌空氣的方式。260~320mOsm/kg的滲透壓對大多數細胞來說是適宜的。在培養(yǎng)過程需補充足夠的氧氣(一般是通無菌空氣),氧氣供給是細胞大規(guī)模培養(yǎng)的一個限制因素。氧化還原電位適用于大多數細胞的值是+75~l00mv左右。在培養(yǎng)過程中,還要補充一些成分,如轉鐵蛋白、胰島素、谷氨酰胺等。
二、大規(guī)模細胞培養(yǎng)的工藝類型
無論呈懸浮生長的細胞還是貼壁生長的細胞,按操作方式,將工藝類型可以分為以下四種:
1.分批式培養(yǎng) 星指將細胞和培養(yǎng)物一次性加入培養(yǎng)反應器內,在細胞生長和產物生成同時進行,經過一段剛司反應后,將整個反應體系取出。這種培養(yǎng)方式,細胞生長的環(huán)境處在不斷變化之中,如營養(yǎng)物質不斷減少,乳酸等代謝抑制物增加,不能使細胞處在一個**優(yōu)化的條件下,因此在應用過程中受到一定的限制。
2.流加式培養(yǎng) 針對分批式培養(yǎng)的缺點,在反應過程中不斷加入新鮮的培養(yǎng)液,使細胞繼續(xù)生長繁殖和生成產物,直到反應結束后取出反應體系。它可以避免細胞在代謝過程中的產物以及某些營養(yǎng)物質的缺乏對細胞的抑制作用。#p#分頁標題#e#
3.半連續(xù)式培養(yǎng) 也稱反復分批式培養(yǎng),是指在分批式培養(yǎng)過程中,不斷取出培養(yǎng)物,每次補充以新的培養(yǎng)液,再進行分批式操作。它與流加式培養(yǎng)的區(qū)別是,流加式培養(yǎng)的體積是不斷增加,而半連續(xù)培養(yǎng)的體積是保持不變的。
4.連續(xù)式培養(yǎng) 是指將細胞和培養(yǎng)液一起加入反應器后,采用灌注培養(yǎng)法,連續(xù)排出用過的培養(yǎng)基,與此同時連續(xù)地加入新鮮的培養(yǎng)液,一般不輸出細胞,使細胞在一個恒定、優(yōu)化的環(huán)境下生長和生成產物,但細胞在數量有波動。如果同時取出與培養(yǎng)液等量的細胞則稱為連續(xù)–流動式培養(yǎng),它是在真正內環(huán)境上保持穩(wěn)定,營養(yǎng)物質、代謝產物和細胞數量不存在波動。這種方法適用于懸浮細胞和在微載體上生長的細胞。
在這些工藝中,以連續(xù)培養(yǎng)為**佳類型,因為系統(tǒng)優(yōu)化的環(huán)境符合細胞的生理和代謝規(guī)律,有利于細胞的生長、增殖和生成產物,但也有些缺點,如培養(yǎng)基的消耗量大,操作過程復雜,增加了污染機會等。
三、 大規(guī)模細胞培養(yǎng)方法
培養(yǎng)方法比較多,大致可以分為懸浮培養(yǎng)、固定化培養(yǎng)和微載體培養(yǎng)法。
1.細胞懸浮培養(yǎng)法 細胞在培養(yǎng)液中呈懸浮狀態(tài)生長繁殖的培養(yǎng)方法稱之為懸浮培養(yǎng)法。適用于培養(yǎng)細胞株、腫瘤細胞、血液細胞及淋巴組織細胞,用于大量生產疫苗、α–干擾素、白介素及McAb(單克隆抗體)等藥品。但此法不適用于少數轉化細胞和人二倍體細胞(WI-38、MRC-5),這些細胞在這種條件下培養(yǎng)則完全不能生存。一些原本貼壁生長但具有懸浮培養(yǎng)潛能的細胞在體外經誘導和選擇后,可以應用此種體系進行培養(yǎng),如由L929細胞誘導出的LS細胞系,由HeLa細胞誘導出的HeLa-S3細胞等。
此種體系明顯的優(yōu)點是培養(yǎng)的體積大、成本低,可連續(xù)收集部分細胞進行移植傳代培養(yǎng),傳代時無需消化分散,免遭酶類、EDTA及機械損害。細胞處于均勻一致的培養(yǎng)基中,因此可以獲得穩(wěn)定狀態(tài),并且容易放大。細胞回收率高,并可連續(xù)測定細胞濃度,還有可能實現大規(guī)模直接克隆培養(yǎng)。
為了確保細胞在培養(yǎng)過程中呈單顆粒、均勻懸浮狀態(tài),在培養(yǎng)系統(tǒng)中使不同的培養(yǎng)相之間(生物學的、液體的、氣體的)保持合適的物質轉移率,以及便利散發(fā)系統(tǒng)產生的熱量,需采用通氣攪拌或空氣提升式(常簡稱氣升式)生物反應器。
通氣攪拌生物反應器,其裝置中的攪拌器(圖13-l所示)具有籠式的通氣腔和消泡腔。氣液交換在由200目(75μm)不銹鋼絲網制成的通氣腔內進行,在通氣過程中所產生的氣泡經管
道進入液面上部的消泡腔內,氣泡碰到鋼絲網,破裂分為氣體和液體兩部分,從而達到了深部通氣和避免產生氣泡的目的。
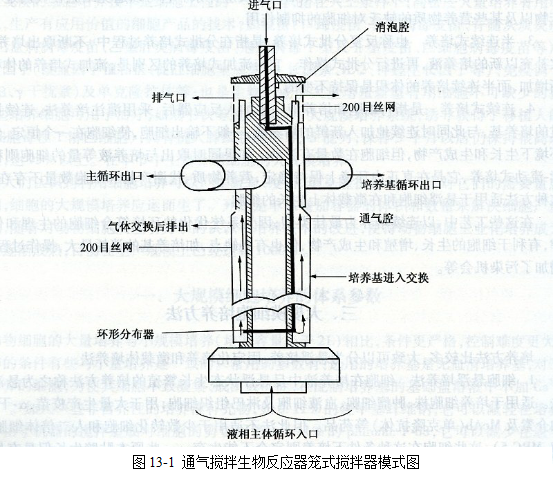
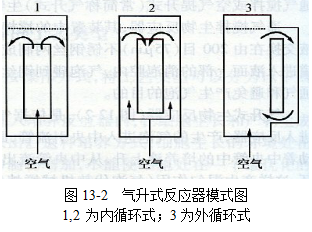
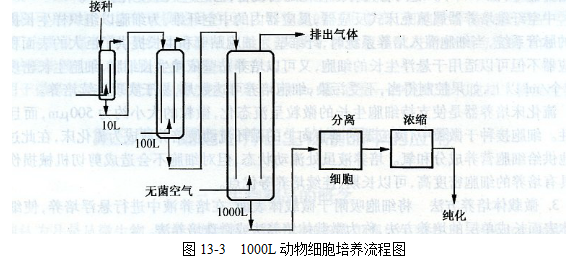
氣升式生物反應器(圖13-2),是依據氣泡柱的原理設計的,氣體混和物從底部的噴射管進入反應器,產生的氣泡進人中央引流管,此時管內的培養(yǎng)液的密度將小于外周的培養(yǎng)液,推動著中央管中的培養(yǎng)液上升,從中央管流出的培養(yǎng)液向下循環(huán)到容器的外側,從而形成一個循環(huán),這樣產生混勻作用(氣泡代替機械攪拌細胞),與此同時進行供氧。這種反應器有2種類型,內循環(huán)式和外循環(huán)式,一般采用的是內循環(huán)式,也有采用外循環(huán)式。目前10 000L的氣升式反應器已經設汁成功并投入使用。圖13-3所示是1000L動物細胞氣升式培養(yǎng)流程圖。體外懸浮液細胞密度一般在5×106個/ml以下,要提高細胞產量,需擴大細胞培養(yǎng)規(guī)模,規(guī)模越大則越難控制。
2.固定化培養(yǎng)法 將細胞限制或定位于特定空間位置的培養(yǎng)技術稱之為細胞固定化培養(yǎng)法。動物細胞幾乎皆可采用固定化方法培養(yǎng)。固定化方法有吸附法和包埋法。
吸附法是**簡單的固定化培養(yǎng)方法。細胞在適當的條件下能夠貼附在載體(如陶瓷顆粒、玻璃珠及硅膠顆粒)的表面,或附著于中空纖維膜及培養(yǎng)容器表面。由于其負載能力不高,有時細胞會從載體表面脫落,不能達到保護細 胞的目的,同時細胞的擴散沒有限制。
包埋法是將細胞包埋于多聚物(蛋白質、碳氫化合物)等海綿狀基質中的進行培養(yǎng)方法。蛋白質多聚物有明膠、膠原和纖維蛋白質。明膠在30℃以上即融化,而細胞的**適生長溫度是37℃,所以不能作為動物細胞包埋的介質。可溶性纖維蛋白原通過凝血作用后,生成不可溶性纖維蛋白,但機械穩(wěn)定性差。但如果這種轉化作用在兩相系統(tǒng)中完成,則機械強度增加,形成球形,此多聚物適合用于包埋貼壁依賴性生長的細胞。碳氫化合物的多聚物可以用于包埋細胞的有海藻酸鈉和瓊脂糖。海藻酸鈉與細胞混和后,當加入一定濃度的氯化鈣時,能形成凝聚的小球,即不溶的海藻酸鈣,細胞即包埋在小球內。這種方法可以用于包埋血細胞。**后用檸檬酸鈉或離心破碎聚合物,收獲細胞和產物。許多瓊脂糖都適用于動物細胞的固定化,它需借助于石蠟油的作用形成小球體(80~ 200μm)。這種包埋方法**適合于懸浮細胞,被廣泛用于培養(yǎng)雜交瘤細胞產生單克隆抗體。#p#分頁標題#e#
固定化培養(yǎng)優(yōu)點在于,細胞可維持在較小體積培養(yǎng)液中生長,可以獲得較高的生長密度((50~200)×106個細胞/m1),細胞損傷程度低、培養(yǎng)的壽命長,易于更換培養(yǎng)液,細胞和培養(yǎng)液易于分離,培養(yǎng)液中產物濃度高,簡化了產品分離純化操作。
這種方法所采用的生物反應器有螺旋卷膜培養(yǎng)器、多層托盤式培養(yǎng)器、中空纖維及流化床式培養(yǎng)器等(圖13-4)。后兩者具有工程化配套設備,已經進入工業(yè)化生產。
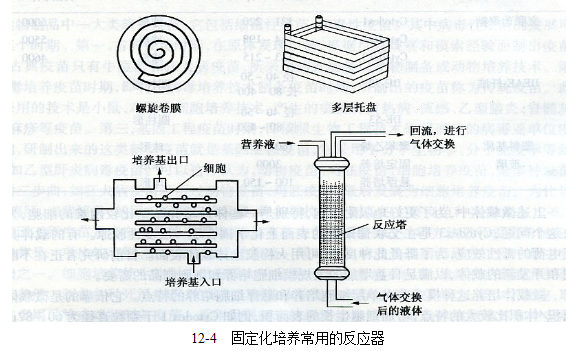
中空纖維培養(yǎng)器屬填充床式反應器,反應器內的中空纖維,為細胞以組織樣生長提供了復雜的脈管系統(tǒng),當細胞灌人培養(yǎng)系統(tǒng)時,纖維壁為細胞貼壁和生長提供了巨大的表面積。這種反應恭不但可以適用于懸浮生長的細胞,又可以培養(yǎng)貼壁依賴生長細胞,細胞生長密度可高達108個/ml以上,如果控制得當,不受污染,細胞培養(yǎng)可達數月,易于實現連續(xù)培養(yǎng)。
流化床培養(yǎng)器是使支持細胞生長的微粒呈流態(tài)化,微粒的大小約為500μm,而且具有多孔性。細胞接種于微粒中,反應器的垂直向上的循環(huán)流動使培養(yǎng)液成為流化床,在此過程中不斷地供給細胞營養(yǎng)成分和氧。培養(yǎng)液雖處流動狀態(tài),但對細胞不會造成剪切機械損傷。所以它具有培養(yǎng)的細胞密度高,可以長期、連續(xù)培養(yǎng)等優(yōu)點。
3.微載體培養(yǎng)方法 將細胞吸附于微載體表面,在培養(yǎng)液中進行懸浮培養(yǎng),使細胞在微載體表面長成單層的培養(yǎng)方法,稱為微載體培養(yǎng)法或微珠培養(yǎng)法。
動物細胞貼附在微載體表面生長與細胞表面及微載體表面的化學–物理性質有關,微載體表面帶有正電荷,而細胞表面帶有負電荷,這種靜電吸引作用使細胞易于在微載體表面貼附,一些細胞分泌纖粘素和冷析球蛋白(兩者都是糖蛋白),起到細胞與載體表面的架橋作用,而溶液中的鈣、鎂離子等二價離子作為糖蛋白結合的媒介。
用于制備微載體的材料的理想條件是對細胞無毒,具有一定的親水性,易于貼附細胞,其密度要略大于培養(yǎng)液,在1.03~1.05之間,但不能大于1.1,能夠高壓滅菌、重復利用,載體的直徑在40~120μm范圍內等。
目前已被選用的材料有交聯萄聚糖、DEAE-纖維、塑料基質、玻璃介質等,如表13-5列出常用的類型和特征。
表13-5 常用的微載體類型
|
基質材料 |
類型 |
直徑大小(μm) |
形狀 |
表面積 |
|
交聯葡聚糖 |
Cytodexl1 |
131~220 |
球形 |
6000 |
上述微載體中,為了更好地吸附細胞,特別是一些體外培養(yǎng)貼壁比較困難的細胞,為了解決這個問題,Cytoder3是在交聯葡聚糖的表面上化學耦合了一層變性膠原。有的載體具有高正電荷的毒性效應,為了降低此種反應,可用火棉膠涂抹載體表面。目前研究者正在不斷地研制和開發(fā)新的載體,以滿足日益增加的大規(guī)模細胞培養(yǎng)和細胞制品的需要。
微載體培養(yǎng)這種模式兼有單層細胞培養(yǎng)和懸浮細胞培養(yǎng)的特點。它依靠的是微載體的表面積/體積比較大的特點,增加細胞生長的表面積,例如Cytodex 1干顆粒直徑為60~87μm,在培養(yǎng)液中可膨脹成160~230μm,每克微粒表面積約為0.6m2,相當于7個標準轉瓶(Φ285×110mm)或6塊多層托盤用的玻璃培養(yǎng)板表面積,細胞生長密度可達105個/ml。通過增加培養(yǎng)罐體積即可達到擴大培養(yǎng)規(guī)模的目的,而且培養(yǎng)基的利用率高,細胞較容易收獲,所占空間小,減少廠房及設備投資,節(jié)約動力消耗及人力,又便于對反應系統(tǒng)進行檢測與控制。由于以上優(yōu)點,貼壁依賴生長的細胞大多采取這種方式。
常用于微載體培養(yǎng)系統(tǒng)的生物反應器是CelliGen罐,它是一種籠式通氣攪拌生物反應器。#p#分頁標題#e#
第三節(jié) 細胞培養(yǎng)在生物制品中的應用
一、生物制品的概念
目前認為凡是從微生物、原蟲、動物或人體材料制備或用現代生物技術、化學方法制成,作為預防、治療、診斷特定傳染病或其他疾病的制劑,通稱為生物制品。狹義的生物制品包括菌苗、疫苗、類毒素、抗毒素和抗血清等。廣義的生物制品還包含抗生素、血液制劑、腫瘤以及免疫病等非傳染性疾病的制劑等。所采用的現代生物學技術,一般認為主要包括基因工程、細胞工程、酶工程和發(fā)酵工程四個部分組成,但還有一些邊緣技術。其中細胞工程包括細胞培養(yǎng)和移植、細胞融合、動物的胚胎工程、植物的微繁殖、單倍體育種、原生質的培養(yǎng)和細胞雜交等。可見在生物制品學中,細胞培養(yǎng)是生物制品的一個主要的應用技術。通過細胞培養(yǎng)所能獲得的生物制品主要有:單克隆抗體、病毒疫苗(狂犬病、乙型肝炎等)、生長因子(表皮生長因子、神經生長因子)、免疫調節(jié)劑(干擾素)、酶(組織血纖溶酶原激活劑)、細胞克隆等。本節(jié)主要介紹細胞培養(yǎng)在疫苗制備中的應用。
二、細胞培養(yǎng)在疫苗制備中的應用
生物制品中一大類就是疫苗,它包括細曲性疫苗、病毒性疫苗。其中病毒性疫苗的發(fā)展可分為三個時期。**,古典疫苗時期,在原體發(fā)現以前,根據反復觀察和摸索經驗而制出疫苗時期,古典疫苗只有牛痘苗和狂犬病疫苗,所采用的方法是以動物制備或動物培養(yǎng)技術。第二,病毒培養(yǎng)疫苗時期,即利用病毒培養(yǎng)技術制備疫苗時期,所制出的疫苗稱為傳統(tǒng)疫苗。此時所采用的技術是小鼠、雞胚和細胞培養(yǎng)技術,產生的疫苗有黃熱病、流感、乙型腦炎、脊髓灰質炎、麻疹等疫苗。第三,基因工程疫苗時期,即依照生物工程技術研制而出的病毒亞單位疫苗時期.研制出來的這類新型疫苗就是基因工程疫苗,它采用了分子生物學、分子免疫學等新技術,如乙型肝炎病毒疫苗。可以這樣認為,動物疫苗、雞胚疫苗、細胞培養(yǎng)疫苗,是多種疫苗發(fā)展的三步曲,如狂犬病疫苗經歷過動物疫苗、雞胚疫苗,**后發(fā)展為細胞培養(yǎng)疫苗。為比較動物、雞胚、細胞培養(yǎng)、基因工程疫苗的發(fā)展,將各種技術所制備的主要疫苗種類列入表13-6。
基因工程疫苗是現在疫苗的發(fā)展趨勢,但有些疫苗仍不能通過這種方法進行疫苗的制備。細胞培養(yǎng)是一項比較可行、簡單制備疫苗的策略。同時細胞培養(yǎng)也是病毒研究工作中**主要的基礎之一。細胞培養(yǎng)使生物制品學有很大的發(fā)展,可以獲得純系病毒,給活疫苗選毒種提供了**佳條件。制備各種死活疫苗,尤其加工制備濃縮提純疫苗,大大提高了效力,減少了反應,取代了許多用動物或雞胚制備疫苗。近年發(fā)展起來的懸浮細胞培養(yǎng)、微載體細胞培養(yǎng)和中空纖維培養(yǎng),已經走上了大批量、工業(yè)化、自動化培養(yǎng)技術。
表13-6 各種技術所制備的主要疫苗
|
疫苗種類 |
主要疫苗 |
制備方法 |
|
動物疫苗 |
疫苗(牛、羊淋巴液) |
雞胚、細胞培養(yǎng) |
在疫苗制備上,細胞培養(yǎng)相對于動物培養(yǎng)、雞胚培養(yǎng)仍有許多得天獨厚的優(yōu)點。
1.細胞沒有特異性的免疫力。細胞在離體組織培養(yǎng)后,不存在免疫作用,易被病毒感染。
2.病毒敏感范圍廣泛,有些病毒具有嚴格的宿主及組織特異性,但離體的細胞培養(yǎng),對病毒的敏感范圍就比較廣泛。如脊髓灰質炎病毒可以在非神經細胞上生長,對原始人羊膜細胞不敏感,但在原代培養(yǎng)的羊膜細胞則敏感,并有細胞病變。腸道病毒、呼吸道鼻病毒等大都能在猴腎細胞培養(yǎng)上生長。
3.在分離病毒時,細胞培養(yǎng)可大量接種標本,從而增加了分離幾率。
4.提高了收獲物的純度,易于加工處理。
5.細胞培養(yǎng)瓶間的差異比較小,大大地提高廠實驗的準確性、重復性。
目前常用的細胞培養(yǎng)的細胞類型及所制備的疫苗類型如表13-7所示。
#p#分頁標題#e#表13-7 細胞培養(yǎng)制備疫苗所用的細胞類型和疫苗類型
|
細胞類型 |
疫苗 |
|
人成纖維細胞 |
HAV(甲型肝炎病毒) |
下面以狂犬病疫苗的制備為例簡單介紹苗的制備過程。
1.使用12g左右健康的金黃地鼠,無菌取腎,去除腎包膜、結締組織及血凝塊。
2.將腎皮質切成1mm3大小的組織碎塊,丟棄髓質部分。
3.用0.25%的胰酶消化組塊30分鐘。
4.離心收集細胞,并制成單細胞懸液。
5.按常規(guī)培養(yǎng),制成單層細胞。
6.接種病毒,用10L轉瓶繼續(xù)培養(yǎng),分兩階段進行,37℃培養(yǎng)3天,于第4天收集病毒溶液;更換培養(yǎng)液,33℃繼續(xù)培養(yǎng),3天后,收集病毒培養(yǎng)液。
7.進行病毒毒力滴定試驗,要求達到5LogLD50/ml。
8.經過0.45μm濾膜濾過,用分子量30萬超濾膜進行超濾濃縮,濃縮后用1︰1000 ~ 1︰10 000的9-丙內酯滅恬,滅活后的濃縮液經過凝膠過濾柱層析及離子交換柱層析進行兩步純化試驗。
第四節(jié) 細胞培養(yǎng)在藥物開發(fā)中的應用
人類的進步可以說是與疾病不斷斗爭的過程,而藥物是用于頂防、診斷和治療疾病和按需要有效調節(jié)人體生理功能的重要物質,是人類防病治病、提高人體健康水平的有力武器。所以科學家們利用各種技術研制開發(fā)治療疾病的藥品。尤其是現在,在藥物開發(fā)上有了很大進步,如以基因工程、細胞工程、酶工程、發(fā)酵工程為主體的現代生物技術和組合化學技術,已經成了藥物開發(fā)的一個技術支柱。正是由于這些技術的進步,并應用這些技術改造傳統(tǒng)的制藥工業(yè),使得開發(fā)出的藥物具有高效、低副作用等優(yōu)點。
一個藥物的開發(fā),包括以下幾個過程:疾病流行趨勢、市場需求、技術水平現狀等基本情況調查;開發(fā)項目立題論證,視制藥公司的人力、財力、設備等綜合實力而確定立項研究;開展包括篩選、合成、提取、發(fā)酵等創(chuàng)新藥物的研究;創(chuàng)新藥物理化性質及其化學結構的研究,動物篩選試驗,發(fā)現候選化合物;開展包括藥效藥理、一般藥理、一般毒性、特殊毒性、代謝、工藝與制劑研究等的新藥臨床前試驗研究;開展包括I、Ⅱ、Ⅲ期試用的新藥臨床試驗;申請承認許可,新藥上市。調查結果表明,在20世紀90年代,一個新藥開發(fā)成功,需15年,臨床前期試驗需要6.5年,I、Ⅱ、Ⅲ期臨床分別需要1.5年、2年、3.5年,美G食品與藥品管理局(FDA)審批需1.5年。一般有這樣的規(guī)律,在每5000 ~ 10 000個進入臨床前期試驗的化合物中,只有5個進入臨床試驗,到**后只有1個獲得批準上市,整個過程成奉需要花費5億美元。由此可以看出,一個新藥的開發(fā),是一個風險大、周期長、高投入的過程。
在當今世界,科技發(fā)展迅猛,在不斷探索新工藝、新方法、新設備來解決開發(fā)藥物中的周期長、成本高的問題。由于有大量的化合物進入臨床前期的藥理學、毒理學、藥效學等研究和篩選。在這個篩選過程中,根據所選用的材料和藥物的作用對象以及操作特點,可將篩查模型分為三種:整體動物水平、組織器官水平、細胞分子水平。目前得到廣泛應用的就是細胞分子水平的藥物篩選模型。它與整體水平、器官組織水平的模型相比,細胞系的生物學特征較為一致,可用于觀察藥物對細胞形態(tài)及生理特征的影響,從而判斷藥物療效及其毒性,具有材料用量少、藥物作用機制比較明確、可以實現大規(guī)模篩選、縮短新藥篩選周期等特點。細胞分子水平藥物篩選模型的應用為自動化操作奠定了基礎,使藥物篩選由傳統(tǒng)手工篩選形式轉變?yōu)橛捎嬎銠C控制的自動化大規(guī)模篩選的新技術體系,形成了高通量藥物篩選(high throughut screening,HTS)。采用細胞分子水平篩選模型進行藥物篩選,在兩方面表現出極大的優(yōu)勢為,一是大樣本量的篩選,由于藥物篩選是對未知的探索和發(fā)現的過程,只有擴大篩選對象和范圍,才能找到高質量的藥物。二足實現了一藥多篩,由于這類藥物篩選模型所需樣品很少,可以使珍貴的藥物在多個模型進行篩選,不但擴大了新藥的范圍,而且有助于從老藥中發(fā)現新的用途。據報道,20世紀90年代初期,一個實驗室采用傳統(tǒng)的方法,借助20余種藥物作用靶位,一年內僅能篩選75 000個樣品;到了1997年HTS發(fā)展的初期,采用100余種靶位,每年可篩選1 000 000個樣品;而到1999年,由于HTS的進一步完善,每天的篩選量就高達100 000種化合物,因而,稱之為超高通量篩選(ultrahieh-throughput screening)。這種新的飛躍,將大大加速新藥發(fā)現的速度。#p#分頁標題#e#
組織細胞培養(yǎng)技術的進步為高通量藥物篩選奠定了技術基礎。這是組織細胞培養(yǎng)技術在藥物開發(fā)應用中的一層含義,另一層含義是利用細胞培養(yǎng)制出新的藥品,這一部分主要是指利用基因工程和細胞工程獲得(在前面有介紹)。在本節(jié)中主要介紹細胞培養(yǎng)技術在藥物篩選測試中的應用。
培養(yǎng)的細胞可以用于藥物高通量篩選,但亦一些不足:①由于在體內存在著許多因素可能對藥物進行修飾,導致藥物作用的增強或減弱,由無致癌性轉變具有致癌性,所以在細胞培養(yǎng)條件所得的結果可能不一定就適用于體內。②藥物對細胞的作用可以有多種表現,但由于檢測手段和檢測方法的限制,可供藥物篩選的指標有限,所以在進行測試時應盡量取多種細胞類型,擴大檢測范圍。
藥物測試的基本程序如下:
1.細胞選擇 利用細胞培養(yǎng)技術進行藥物測試時,shou先根據目的選擇合適的細胞類型,如果選擇的細胞不合適,則影響實驗結果的可信性和參考價值。目前用于藥物開發(fā)測試研究的培養(yǎng)細胞主要來源于原代細胞、細胞系和細胞株,這三種細胞培養(yǎng)的特點見表13-8。
表13-8 三種培養(yǎng)細胞的某些特征
|
|
原代細胞 |
細胞系 |
細胞株 |
|
培養(yǎng)生存時間 |
數小時–數天 |
數月–無限期 |
常為無限期 |
從表中可以看出,這三種細胞有各自的優(yōu)缺點,在應用時要根據具體情況進行選擇。
如果在藥物效應不明的情況下,對實驗組細胞的選擇可不必十分嚴格,但為了解藥物可能的作用,**好多選用幾種類型的細胞進行。如已知藥物有特殊效應的或欲求獲得對某一類細胞產生作用時,應盡量采用相應的細胞。如果是神經系統(tǒng)藥物則用神經細胞,抗癌藥物用癌細胞,如HeLa、KB、HL–60(人急性早幼粒白血病)、U937(人單核細胞白血病)、A-549(人肺腺癌)等,這樣獲得的結果才有參考價值。
對照細胞是實驗組合中不可少的組成部分。原則上實驗細胞和對照細胞越相似越好。如檢測某一藥物用的是正常細胞,對照細胞可用同一細胞但不用藥物處理或僅用溶媒液處理即可。
2.藥物的溶解和保存 藥物在具體使用前一般都需配成貯存液,要根據藥物的性質選擇適當的溶劑,如果是水溶性,即可用不完全培養(yǎng)基或者PBS液配制100×母液,用時按所需濃度進行稀釋。有些藥物不適宜水溶液配制的,可以用DMSO(二甲基亞砜)或無水乙醇進行配制,要求要藥物工作濃度下,DMSO和乙醇的濃度要小于0.2%(V/V),這時不會對細胞產生毒性,進行實驗時,對照組要加入同等量的溶劑。所配制的母液根據藥物的性質進行保存,一般在-20℃.但有的藥物需放置于-70℃比較穩(wěn)定,一些光敏藥物還需避光保存。
3.藥物活化 運用培養(yǎng)細胞進行藥物檢測有時并不能反應體內情況,確些藥物經過肝臟后,會發(fā)生生物轉化作用,如可能會使得原來沒有細胞毒性的藥物會產生毒性。為了彌補此缺陷,設計了一個與體內環(huán)境比較相似的藥物活化試驗,運用大鼠或人的肝細胞恬檢材料和S9混和液在體外將藥物活化。受試藥物和S9混和液與細胞接觸作用時間一般為6小時。
4.藥物劑量 在試驗開始前,需先確定待測藥物適用劑量。與利用動物做試驗一樣,先宜測出半效應量(median infective dose,LD50)。它的計算方法有兩種方法,一種是粗略估計法,另外是借用藥物的LD50(半致死量)進行精確計算。如果被測藥物是抗癌藥物或者是三致物(致畸、致突變、致癌)則兩者可等同。
粗略估計法:根據藥物的作用選擇合適的指標進行藥物的有效反應性。例如檢測抗癌藥物殺細胞活性,如果其已在臨床應用或做過動物試驗,此時可按已用藥量推算。在無據可依的情況下,只有按試驗者經驗確定一組梯度數值,如11μg/ml、3μg/ml、5μg/ml、10μg/ml、20μg/ml然后安排一組培養(yǎng)細胞,共2瓶/組×5。分別向五組培養(yǎng)細胞中加入五個劑量的藥物,作用一定時間后,用臺盼藍染色法測試死活比數(存活的細胞可以排斥許多染料,如臺盼藍,細胞不著色,而死亡的細胞和嚴重受損細胞由于細胞膜的通透性改變,染料可通過細胞膜而被著色)。將所得數值進行直線回歸,取50%死亡細胞時的濃度做為粗ID#p#分頁標題#e#50。亦可用MTT的方法,設立一個對照組,不加入藥物,以此為100%的存活,取OD570的值下降到50%的藥物濃度。這種方法在96板上即可完成,方便、簡單,比較準確。比較適用于貼壁細胞,如果足懸浮細胞,在操作過程中需要用到96孔板離心機。
借用計算LD50一些常見的方法有:寇氏法(Karber’s method)、改良寇氏法和何爾恩法(Horn’s method)等,這些方法比較準確。在計算過程有些復雜的運算,為了解決這個問題,一些研究機構開發(fā)出計算機軟件,使得計算更準確和快速,如Bliss法。下面的計算公式是改良寇氏法:
LogID50=Xm-i (∑P-0.5)
i=Log (**大劑量/相鄰劑量)
∑P=各組陽性反應率之和
此法比較簡單,準確性也較高,但要求**大陽性反應組的陽性率>80%,而且**小反應陽性率不小于20%,否則要換用其他方法。
待ID50確切值獲得后,取ID50/10作為主要測試劑量。
5.藥物作用時間 確定出待測藥物的ID50后,取ID50/10為試驗劑量。根據細胞的生長特性進行加藥。如原代培養(yǎng)的細胞或收獲的貼壁細胞(經胰酶消化過)一般要等到**少12小時以后,此時細胞對藥物的敏感性恢復到非胰酶水平。一般的**適宜時間是在接種后約第48小時,此時細胞已進入指數生長期,加藥后可再更換一次培養(yǎng)液,然后加入測試藥,置溫箱中培養(yǎng)。藥物在瓶皿中于細胞接觸時間要依據藥物的性質(如半衰期)和細胞的敏感性決定,在試驗開始時,需進行預實驗,做出藥物的作用效應和時間關系的曲線,依此進行選擇適當的作用時間。一般不應少于8小時,也可處理12~24小時或更長些,然后棄掉含有藥物的營養(yǎng)液,用不完全培養(yǎng)液洗1~2次,再補充新的培養(yǎng)液繼續(xù)培養(yǎng)。
6.細胞的觀察 在試驗開始后,每天或按一定時間間隔,取出1組培養(yǎng)細胞進行觀察,從瓶皿中抽出蓋玻片做各種方法的染色觀察,瓶皿中余下細胞制成細胞懸液通過計數等手段檢測細胞數量。總之為檢測藥物對細胞生物效應,可結合應用各種技術方法,進行有針對性的檢測。
通過一些觀測指標對細胞進行觀察,才能了解藥物對細胞的效應如何。各種藥物對細胞的生物效應各不相同,必須選擇相應的指標。下面列出了一些常用的檢測指標。
(1) 細胞形態(tài)(光鏡下):殺傷性藥物作用細胞后,可能有多方面反應,一是細胞的形態(tài)和生長變化,在顯微鏡下容易判定,因此這項檢測可作為與臺盼藍檢查細胞同時并舉的指標。有些細胞的形態(tài)是生長的一個特征,如神經細胞在體外生長,可以明顯地觀察到長長的軸突,當用殺傷性藥物作用時,它的軸突會縮短。另一種是對細胞代謝和基因表達的變化。根據形態(tài)變化和生長速率變化的結果,可大致分為以下5個等級:
0度:細胞在一定劑量一定時間藥物作用下,無任何反應,細胞形態(tài)正常,生長能力無改變,貼壁細胞緊貼瓶底,無脫落,細胞的折光性好,以(-)表示。
1度:細胞生長速度減慢,細胞分裂指數下降,細胞輪廓增強,有少量貼壁細胞開始脫落,胞質中出現顆粒,以(+)表示。
2度:細胞生長非常緩慢,細胞分裂指數顯著下降或近于消失,細胞輪廓增強,胞質粗糙,部分細胞脫離瓶底,胞內有顆粒堆積,以(++)表示。
3度:細胞生長完全停止,分裂象消失,細胞回縮,相互間隙加大,細胞輪廓非常明顯,大部分細胞脫落,胞質極度粗糙,內充滿顆粒堆積,以(+++)表示。
4度:細胞全部脫落死亡,殘余的細胞亦瀕死或崩潰溶解,培養(yǎng)基的顏色顯示為堿性,以(++++)表示。
(2) 化學成分檢測:有的藥物對細胞功能的作用常表現在對某種物質代謝產生抑制作用(如膠原),則以測試該成分在細胞內或培養(yǎng)液中的含量,即可感知藥物的效應。對酶有作用時,可用組織化學方法或電泳等方法檢測。TorranceCJ等利用現代分子生物學技術,開創(chuàng)了一類針對產物檢測的新的策略,它的檢測速度快、自接,而且在缺少對照細胞的情況下也可進行。作者將一具有K-ras基同突變的結腸癌細胞系DLD-1,通過同源重組的方法將其轉變?yōu)闊oK-ras突變的細胞系.將DLD-l轉染一種黃色熒光蛋白,而將缺失K–ras突變的細胞系轉染有藍色熒光蛋白,二細胞系在不加任何抑制物共同培養(yǎng)時則生長速度相同。研究者用此體系去篩選藥物,在藥物的作用下,如果黃色熒光增強,說明藥物抑制了缺失K-ras突變的細胞的生長,如果藍色熒光增強,則說明藥物抑制了含K-ras突變的細胞的生長。運用這種方法,一共篩選了30 000個化合物,發(fā)現一個胞苷類似物在體外具有抑制含K-ras突變的細胞的生長。
(3) 超微結構的變化:細胞受藥物作用后,形態(tài)上很容易發(fā)生改變,如成纖維細胞突起回縮,上皮細胞相互分離變成梭形等,在一般光學顯微鏡下很容易看到,但這些變化十分不可靠,應主要以超微結構觀察到的改變?yōu)橐罁鐚毎ぁ⑽⒔q毛、內質網、線粒體、細胞核等微細結構的影響。
(4) 細胞死亡檢測:致死效應是藥物檢測中,特別是抗腫瘤藥的重要指標。細胞死亡是一個很復雜的生命過程,shou先應明確細胞死亡的概念。細胞崩潰、脫壁、溶解等是在顯微鏡下可觀察到的細胞死亡現象,同樣也不能完全僅依靠這些為依據,否則一些不能迅速發(fā)生效應的藥物作用易被忽視。應該是凡能引起細胞內相互制約系統(tǒng)中一個或多個環(huán)節(jié)的紊亂,**終導致細胞功能障礙或細胞生命活動不可逆停止的現象,都應視為細胞死亡的范疇。如氰化物抑制細胞氧化磷酸化阻斷ATP供應,氯霉素干擾蛋白質合成抑制細胞修復等,進一步發(fā)展,終會導致細胞死亡。#p#分頁標題#e#
研究細胞死亡的方式對研究藥物作用于細胞的機制是非常重要的。細胞死亡的方式根據病理上的分類有兩種形式,一種是細胞壞死,另一種是細胞凋亡,這是指一種有秩序、受控制并按某種預定程序發(fā)展的生理性自然死亡過程(參見第十一章)。有的藥物可能只通過其中一種形式,而有的藥物作用機制可能二者兼而有之。下表列出了二者的一些區(qū)別(表13-9)。
表13–9 細胞壞死和細胞凋亡的比較
|
|
細胞壞死 細胞凋亡 |
|
細胞形態(tài)變化 |
細胞腫脹,細胞質內出現顆粒,核膜破 細胞皺縮,染色質凝聚,質膜小泡,凋亡 |
(5) 細胞增殖和存活測定:前面提到的用臺盼藍染色觀察和計算細胞的生長速率,不能反應細胞的增殖情況。如若精確測定細胞的增殖和存活,則需進行克隆形成率(反映細胞的存活率)和克隆大小測量(反應細胞的增殖情況)。
(6) 藥物遺傳毒性測試:反映化學藥物的遺傳毒性的檢測可以分為四類:DNA損傷、基因突變、細胞遺傳學改變(染色體畸變和姐妹染色單體互換)、細胞轉化。這些檢測呈對一個藥物的常規(guī)測試。
1)對DNA的損傷檢測
方法一:
【原理】
DNA鏈的斷裂呈化學物對DNA造成損傷的一種常見形式。其常作的檢測方法是DNA解旋的熒光測定(fluorescntric assay of DNA unwinding,FADU)。其原理是:DNA在變性液里會發(fā)生DNA解旋,如果DNA的分子存在鏈的斷裂,這種解螺旋的速度會加快,此時溴化乙錠(EB)在堿性溶液中特異性地嵌入DNA中而引起熒光值增加。通過測定熒光Qd值,即可檢測DNA斷裂情況。
【操作】
①細胞懸液制備:用常規(guī)方法制備細胞懸液,使其細胞濃度為1×106個/ml。實驗分為對照組、實驗組,對照組設T(未變性)、B(空白)和P(部分變性)三組,實驗組只設Pi(部分變性)組,每組三個平行管,每管0.6ml。
②每管細胞中加入0.6ml裂解液(9mmol/L尿素,10mmol/L NaOH,2.5mmo/L EDTA,0.3% Sarcosyl),待15分鐘后細胞裂解,T組加1.2ml抗變性液(1mol/L葡萄糖,14mmol/L巰乙醇),各管沿管壁小心加入變性液I(0.4體積裂解液溶于0.2mol/L NaOH)和變性液Ⅱ(0.4體積裂解液溶于0.2 mol/L NaOH)各0.3ml。然后B管立即超聲處理(功率20%)20秒。變性溫度和時間為0℃,30分鐘,15℃,30分鐘,變性時需避光進行。B、P和Pi組各加入溴化乙錠液(1μg/L EB溶于13.3mmol/L NaOH),室溫下MPF-4熒光分光光度計熒光測定,條件為激發(fā)光波長520nm,熒光波長590nm。
③由T、P、B管熒光值計算殘存雙鏈DNA百分數(D):
D=[(P—B)/(T—B)]×100%
為獲得直線型劑量效應關系曲線引入Qd值:
Qd=100(1gPo-lgPi)
Qd值大小反映廠DNA鏈斷裂程度的高低,值越大表明斷裂越多,式中的Po為未處理細胞D值,Pi為處理的細胞D值。
方法二:
【原理】
當染色體DNA斷裂時,分子量與完整的基因組DNA相比比較小,通過離心可與之分離用熒光物質Hoechst33258染色,計算DNA的斷裂的比率。
【操作】
①1000g離心收集細胞,細胞沉淀用lml裂解液(10mmol/L Tris-Cl,pH7#p#分頁標題#e#.5,1mmol/L EDTA,0.2%TritonX-100)裂解2分鐘。
②13 000g離心20分鐘,含有斷裂的DNA存在上清里,轉移上清**一新的管中;而完整的DNA存在沉淀中,用超聲破碎。
③向兩部分溶液中分別加入等體積的lμg/ml Hoechst33258,37℃染色20分鐘。
④于熒光分光光度汁檢測,激發(fā)光波長360nm,熒光波長450nm,計算斷裂DNA占總DNA的百分數。
2) 基因突變的檢測;
【原理】
人類和嚙齒類動物的正常細胞內X染色體上含有次黃嘌嶺—鳥嘌呤磷酸核糖轉移酶(HG-PRT),它能代謝嘌呤類似物6–硫代鳥嘌嶺(6-TG)或8–氮雜鳥嘌呤(8-AG),形成一種致死性的核苷-5’磷酸鹽,從而殺死正常細胞。在致癌物或致突變物的作用下,細胞X染色體上控制的HGPRT結構基因發(fā)生突變,不能再產生HGPRT,從而使細胞對6-TG具有抗性,這些細胞能夠在含有6-TG的培養(yǎng)液中長成集落。凡能引起堿基替換、移碼突變、缺失和基因重組的誘變劑,均能引起HGPRT基因位點突變,這種突變是不可逆的。已經廣泛用于輻射劑量估算、遺傳毒理學誘變機制等研究。
【操作】
①正常細胞的常規(guī)培養(yǎng),在培養(yǎng)液中加入測試藥物,在進行實驗時要設立對照組,包括陽性對照(可以用苯并芘)和陰性對照組,而且藥物要有濃度梯度。
②37℃培養(yǎng)30小時,加入6μl/ml細胞松弛素B,繼續(xù)培養(yǎng)42小時。
③棄去培養(yǎng)液,用PBS洗2次。
④細胞懸液經冰醋酸:甲醇(1︰3)固定。
⑤制片,Giemsa染色,顯微鏡下計數雙核和多核細胞數。
⑥計算HGPRTJI基因突變變異子數,即含6-TG培養(yǎng)的1000個細胞中雙核或多核細胞數除以不含6-TG培養(yǎng)的1000個細胞中雙核細胞數或多核細胞數。其結果反映細胞HGPRT基因損傷情況。基因突變的劑量–效應關系,隨致突變劑劑量增加而增加。
3)染色體畸變試驗:
【原理】
用于染色體畸變檢測的細胞有人和動物的末梢血淋巴細胞,細胞系有中G倉鼠卵巢細胞(CHO)、中G地鼠肺成纖維細胞(V79)和中G倉鼠肺細胞系(CHL)、人胚肺2倍體成纖維細胞,**常用的是外周血淋巴細胞和CHL,在我G新藥的染色體畸變試驗推薦的**細胞為CHL,其染色體為25條。外周血中的小淋巴細胞幾乎都處于細胞周期的G1和G0期,一般條件下不會再分裂,當培養(yǎng)物中加入PHA,在37℃下,經52~72小時的培養(yǎng),淋巴細胞開始轉化,進入細胞增殖周期,此時可獲得大量的有絲分裂的細胞。再經過秋水仙素處理,低滲、固定后在顯微鏡下可以觀察到良好的中期染色體分裂象。當電離和化學有害物質作用時均可引起染色體的損傷,且與劑量呈良好的線性關系。
【操作】
①用淋巴細胞分離液分離淋巴細胞,常規(guī)細胞培養(yǎng),把細胞分為五組,即陽性對照組、陰性對照組和三個劑量組。**高劑量和**低劑量相差10倍,中間再設一個劑量,陽性對照物為已知的染色體斷裂劑,如絲裂霉素C,劑量為0.02μg/ml;黃曲霉素毒素B,濃度為10-6mol/L等。每組設三個平行樣品。
②收集細胞,加入含有PHA的培養(yǎng)基(如果用CHL細胞則不需加),37℃培養(yǎng)52~72小時。
③加入40μg/ml秋水仙素0.05~0.1ml,終溶度為0.4~0.8μg/ml,37℃培養(yǎng)4小時。
④收集細胞,加入8ml 0.075 mol/L KCI低滲液,制成單細胞懸液,37℃、20分鐘。
⑤離心收集細胞,加入3ml冰醋酸︰甲醇(1︰3)固定細胞,用吸管輕輕吹打后,立即離心收集細胞。加入10ml冰醋酸︰甲醇(1︰3)處理15分鐘。反復操作2次,每次15分鐘。
⑥取出冰預冷的載玻片,每片滴加1~2滴細胞懸液,吹散后,自然干燥后,在顯微鏡下觀察有無細胞分裂象。
⑦Giemsa染色15分鐘,用自來水沖洗殘留液體,干燥后顯微鏡下觀察。進行染色體畸變計數,并攝像。畸變的類型有斷片(F)、雙著絲粒(D)、環(huán)(R)、互換(E)等,電離輻射常見斷片、雙著絲粒、環(huán)等,而化學藥物常見單體斷裂。結果表示為:
總畸變率(%)=(各種畸變類型細胞數/分析總細胞數)×100%
畸變率(%)=(染色體畸變數/染色體總數)×100%
如果采用CHL進行此項實驗,其結果判定是:
|
畸變率 結果 |
畸變率 結果 |
|
<5% 陰性(–) |
20% 陽性(++) |
姐妹染色單體互換試驗
【原理】
當細胞在加有BrdU(5-溴脫氧尿嘧啶核苷)的培養(yǎng)基中進行有絲分裂時,BrdU能取代胸腺嘧啶核苷酸而摻入到新復制的DNA鏈中。在經歷了兩個分裂周期,細胞中期染色體上的兩條姐妹染色單體一條被BrdU半取代,即單鏈取代,另一條的雙鏈都含有BrdU,即雙鏈取代。兩者這種差別可借不同的染色方法將其分別開來,雙鏈都含有BrdU的染色單體在Giemsa染色的情況下染色淺,而一股鏈含有BrdU的染色單體染色深,這種姊妹染色單體上染色深淺相間的現象稱為姐妹染色單體分化現象(SCD)。已分化的兩條染色單體之間如發(fā)生等位點交換的現象,稱為染色單體互換(SCE)。
【操作】
①按常規(guī)細胞培養(yǎng)方法培養(yǎng)CHO細胞,加入受試物,作用2小時(亦可采用外周血細胞培養(yǎng),但在培養(yǎng)過程中加入PHA)。
②棄去培養(yǎng)液,用Hanks液洗滌3次。
③加入含BrdU l0μg/ml的完全培養(yǎng)液5ml,于避光條件下培養(yǎng)24~27小時。
④加入秋水仙素1~2滴(終濃度為0.2μg/m1),繼續(xù)培養(yǎng)4小時后,收集細胞。
⑤按常規(guī)用0.075 mol/L KCl低滲、冰醋酸;甲醇(1︰3)固定,制片。
⑥標本在37℃條件下光化24小時后,置于大培養(yǎng)皿中,上蓋以擦鏡紙,滴加2×SSC液,以溶液不漫沒玻片而能保持標本濕潤為止。
⑦將大培養(yǎng)皿移**60℃水浴箱上,用20W紫外燈距離5cm垂直照射30分鐘。
⑧去掉擦鏡紙,以3%Giemsa染液染色15分鐘,自來水沖洗,晾干,鏡檢。
⑨以每個細胞SCE數計算SCE頻率,每個樣品**少觀察25 ~ 50個細胞。
【注意事項】
BrdU溶液**好現配現用,一次用不完,必須用黑紙(布)避光,4℃冰箱內保存。
BrdU溶液強致突變劑,使用濃度不宜太高,在25μg/ml左右的劑量下對細胞增殖無影響。在培養(yǎng)24小時后加入均可。
用紫外線照射誘發(fā)姐妹染色單體互換時,如果紫外燈功率大,照射時間應相應減少。
經過t顯著性檢驗后,**少有一個劑量組SCE頻率超過對照組2倍,即相當對照組SCE頻率3倍者為強陽性;若三個劑量組超過對照組SCE頻率且有量效關系,井其中**少有一個劑量組與對照組有非常顯著性差異(P<0.001)者為弱陽性,否則為陰性。
5)細胞短期轉化試驗(藥物致癌實驗):細胞轉化是細胞發(fā)生涉及到DNA或基因改變,導致遺傳性狀改變的一種變化。細胞發(fā)生轉化后的性狀可以代代相傳,能夠長期維持和存在。觀察藥物對細胞是否有誘導轉化作用是研究藥物是否有致癌作用的常規(guī)技術。
【原理】
體外培養(yǎng)的細胞在受電離輻射或化學致癌物的作用下,可發(fā)生細胞表型的轉化,包括細胞形態(tài)轉化及生長特性的改變,并形成轉化克隆,而且大量實驗結果表明,細胞形態(tài)學的轉化與體內腫瘤生長有密切的關系。一般細胞轉化試驗的周期需2~3個月,而應用敘利亞金黃地鼠胚胎細胞(SHE)作為靶細胞,經致癌因子作用后第9天即可檢測出細胞形態(tài)的轉化。此試驗模型已廣泛用于環(huán)境致癌物和各種藥物未知致痛性的檢測,成為細胞毒理學中一個基本的實驗方法和手段。
【方法與步驟】
①原代細胞的制備及接種:采用常規(guī)建立細胞系的方法,取妊娠10~20天的敘利亞金黃地鼠的胚胎制備原代細胞懸液,接種細胞為1×106個細胞/ml。每個培養(yǎng)瓶加入1ml細胞懸液,2ml完全培養(yǎng)液及雙抗,37℃溫箱內培養(yǎng)。
②飼養(yǎng)層細胞的準備:直接采用次代培養(yǎng)的SHE細胞制備,每平方厘米生成面積接種3.3×104個細胞,培養(yǎng)基為RPMI–1640或Eagle完全培養(yǎng)液,37℃下培養(yǎng)3~5天。待細胞80%聚集時,進行X射線或60Coγ射線照射,劑量為5.0Gy。照射時細胞培養(yǎng)面朝上,照射后立即用胰蛋白酶消化,然后加入10%小牛血清的完全培養(yǎng)液,調整細胞濃度為2×l04個/mL,接種4×104個細胞(2ml)于一系列25m1培養(yǎng)瓶中。受試物每—濃度組及對照組各12瓶平行樣,置37℃下培養(yǎng)24小時后,即可接種靶細胞。因飼養(yǎng)層細胞受到照射,細胞體積大,不增殖,形態(tài)特異,易與靶細胞區(qū)別開。
也可用地鼠肝細胞制作飼養(yǎng)層細胞,其可使測試系統(tǒng)代謝活化能力提高數十倍**數百倍,而且無細胞毒性。
③靶細胞的制備:取凍存的SHE細胞,作常規(guī)培養(yǎng),當細胞長到80%~90%融合時,即可用胰蛋白酶消化1~1.5分鐘,以完全培養(yǎng)液稀釋**300個細胞/ml,取1ml細胞懸液接種于飼養(yǎng)層細胞培養(yǎng)瓶中,總體積3ml。37%下培養(yǎng)24小時,加入受試物,可設立三個濃度的實驗組,一個陽性對照組(如3-甲基膽蒽,3-MAC或N-甲基-N-硝基-N-亞硝基胍,MNNG),濃度一般為1μg/ml左右。若受試物不溶于水,可先用二甲亞砜溶解,二甲亞砜在培養(yǎng)液中的濃度為0.02%(V/V)。加入受試物后,37℃下連續(xù)培養(yǎng)8天,此時細胞既不傳代,也不換培養(yǎng)液。
④固定、染色:細胞培養(yǎng)**第9天,棄去培養(yǎng)液,用pH6.8磷酸緩沖液將貼壁細胞洗2次,**后加入甲醇3~5ml固定20分鐘,再用Giemsa染液染色20分鐘,**后經雙蒸水沖洗,晾干。
⑤鏡檢:在低倍鏡下觀察細胞轉化克隆的形態(tài)。轉化克隆的成纖維細胞生長無方向性,排列紊亂,形成交叉、旋渦、復層生長(三維空間生長),染色深。正常細胞不形成克隆,或形成的克隆細胞生長有一定方向性,排列規(guī)則,基本上單層生長,染色淺。#p#分頁標題#e#
⑥轉化克隆的判斷:為了便于判斷克隆是否發(fā)生形態(tài)轉化,可將每個克隆劃分為三個區(qū)域。中心區(qū):細胞**致密,常呈三維空間生長,細胞界限不清,非轉化克隆無此現象。外周區(qū):自中心區(qū)邊緣向克隆周邊延伸,在此區(qū)域中細胞單層生長,但排列不規(guī)則,形成交叉、重疊,而正常細胞呈方向性生長。細胞稀疏的區(qū)域:在克隆外周區(qū)之外可有一個細胞群體很稀疏的區(qū)域,在此區(qū)域內,細胞間隙較大,生長較紊亂,細胞突起重疊,但不典型。此區(qū)域中細胞形態(tài)不能作為判定轉化克隆的依據。
⑦判定陽性結果的標準:根據Dumkel等的建議,SHE細胞準化試驗中符合以下標準者即可判定為陽性結果:明確的劑量–效應關系。兩個連續(xù)劑量組中都有2個或2個以上的轉化克隆。單個劑量組中,有3個或3個以上的轉化克隆。
若出現以下情況中,僅屬可疑陽性:單個劑量組中,只出現1個或2個轉化克隆者。在兩個不連續(xù)的劑量組中,出現1~2個轉化克隆者。在兩個連續(xù)的劑量組中,僅出現1個轉化克隆者。
據此尚難做出明確判斷者,需做其他實驗進行確定,將所形成的克隆分離出來,進行染色體數目和核型分析、細胞凝集實驗、半固體瓊脂培養(yǎng)基中集落開形成率測定、動物接種致瘤試驗,以增強結果的可靠性。其中半固體瓊脂培養(yǎng)**為可靠,當動物接種時出現腫物生長并結合病理確診則**有說服力。
【注意事項】
胚胎原代細胞比傳代細胞易轉化,同窩動物的胚胎細胞轉化結果較穩(wěn)定。
小牛血清必須及時滅活,無微生物、支原體污染。活力低的血清或污染血清不易使細胞發(fā)生轉化。
培養(yǎng)液配制保存時間不宜過長,否則會降低細胞轉化率。
**接種的細胞密度應高一些,由于接觸抑制作用,減少細胞倍增次數,可提高轉化率,否則會降低轉化率。
第五節(jié) 細胞培養(yǎng)在臨床中的應用
臨床醫(yī)學是認識和防治疾病、保護和增進人體健康的科學。細胞培養(yǎng)技術在疾病的診斷、治療上有重要的應用。近年來的細胞培養(yǎng)技術的發(fā)展和其他基礎醫(yī)學的理論和技術如分子生物學、物理學、化學等促進了臨床醫(yī)學的蓬勃發(fā)展。
一、細胞培養(yǎng)在臨床診斷中的應用
1.細胞培養(yǎng)對臨床上的病原研究起了很大的推動作用 一些疾病通過細胞培養(yǎng)技術得以發(fā)現和確定病原。臨床上多種致病病原如細菌、病毒、寄生蟲等的確定和藥物敏感試驗是通過細胞培養(yǎng)技術完成的。如臨床上廣泛進行的對呼吸道、泌尿生殖道分泌物的病原菌培養(yǎng)和篩選敏感抗生素。通過培養(yǎng)確認病原的方法,人們也新發(fā)現和診斷了多種以往不能明確的疾病類型,如艾滋病病毒就是通過細胞培養(yǎng)發(fā)現的。使用組織細胞培養(yǎng)技術,可對已知的病毒(如脊髓灰質炎、麻疹病毒、腮腺炎病毒等)進行深入的研究,還有助于發(fā)現大量人類未知的腸道和呼吸道病毒,這些病毒在雞胚和小鼠中并不繁殖。
目前在臨床上應用微量全血培養(yǎng)技術進行珍斷HIV感染,用于兒童HIV垂直感染的早期診斷及可疑感染者的確認。HIV分離培養(yǎng)是當今HIV垂直感染診斷的依據,早期雖然已有常規(guī)培養(yǎng)方法來檢測HIV,但這些技術依賴于血液成分的分離,花時間并難以從兒童得到有效血量。血清學檢測在<24個月齡的小兒中有一定的局限性,其主要原因是因為24個月內的嬰兒體內可能仍存有母體的HIV抗體,而ELISA法不能確定抗HIV抗體是來源于母親還是嬰兒自身產生。為此,Bayliss等人**用微量全血培養(yǎng)法從HIV/AIDS感染者血液中分離出HIV病毒。
2.細胞培養(yǎng)在臨床病因診斷中的應用
(1)造血系統(tǒng)疾病診斷:造血或血細胞生成是一個復雜的、多層次的細胞分化生物過程,
包括造血干細胞的自我更新和分化增殖。其分化增殖產生各系列定向造血祖細胞,后者進一步增殖分化產生成熟的血細胞。造血這一過程需要一個適合的微環(huán)境,又受到局部和全身的生長因子、抑制因子的調節(jié)。造血于祖細胞的研究,可使人們了解造血的正常生理過程,包括其分化、成熟及調節(jié)機制。
正常人骨髓中多能造血干細胞約占細胞總數的0.4%,定向祖細胞約占3%。外周血中的比例更低。由于這些細胞在一般形態(tài)上與淋巴細胞無明顯差別,不能直觀地研究其特性。因此主要通過骨髓和血液細胞體外培養(yǎng)的方法,根據其增殖能力和所形成集落的特征不同,將不同的細胞加以區(qū)分。現在也可以通過象免疫磁珠等方法先進行干祖細胞的純化分離,再進行培養(yǎng)擴增。造血細胞的培養(yǎng)分析則已作為造血干細胞克隆性疾病的診斷和治療效果分析的常用方法。G內外使用的造血細胞培養(yǎng)方法有許多種,按培養(yǎng)基形態(tài)可分為液體培養(yǎng)法和半固體培養(yǎng)法;按半固體培養(yǎng)支撐物的種類可分為血漿凝塊法、瓊脂法和甲基纖維素法;按血清的有無可分為無血清液體培養(yǎng)法和血清半固體培養(yǎng)法等。
造血干細胞可向多種定向祖細胞分化增殖,所以不同類型的造血干細胞克隆性疾病如骨髓增生異常綜合征(MDS)、急慢性白血病、再生障礙性貧血(AA)、骨髓增殖性疾病(MPD)等造血細胞的集落分析均有一定特點。目前已發(fā)展了多種造血細胞的培養(yǎng)體系,即在常規(guī)培養(yǎng)基中應用不同的細胞因子組合用以培養(yǎng)和擴增不同的定向祖細胞。如混合集落形成細胞(CFU-Mix)培養(yǎng)、粒單細胞集落形成細胞(CFU-GM)培養(yǎng)、巨核細胞系集落形成細胞(CFU-MK)培養(yǎng)、紅系祖細胞(BFU-E)培養(yǎng)、成纖維細胞祖細胞(CFU-E)分析和骨髓長期培養(yǎng)等。重癥再生障礙性貧血患者的骨髓、外周培養(yǎng)血幾乎都無集落形成,可認為是CFU-Mix階段障礙;MDS病人的CFU-GM培養(yǎng)常表現為集落減少而集簇增多;AML骨髓粒–單核系祖細胞(CFU-GM)集落不生成或生成很少,而集簇數目增多。#p#分頁標題#e#
(2)與染色體變化相關疾病的診斷:染色體分析亦稱核型分析(karyotype analysis),是在細胞培養(yǎng)的基礎上發(fā)展起來的一項專門技術。目前隨著顯帶技術的應用以及高分辨率染色體顯帶技術的出現和改進,能更準確地判斷和發(fā)現更多的染色體數目和結構異常綜合征,如21三體綜合征等,還可以發(fā)現新的微畸變綜合征。染色體分析標本的來源,主要取自骨髓、外周血、絨毛、羊水中胎兒脫落細胞和臍血、皮膚等各種組織。由于只有處于中期的染色體才能在光學顯微鏡下看到它的獨特結構,因此染色體分析均需用正在分裂的細胞。體內一般只有骨髓細胞含有足夠比例的處于有絲分裂的細胞,其他細胞均需要通過一定條件的培養(yǎng)使之達到分析的要求。外周血淋巴細胞因其容易獲得,也常用于體細胞染色體畸變的細胞遺傳學分析。一般來說,在有分裂原(如植物血凝素、美洲商陸分裂素或伴刀豆球蛋白A)存在的情況下,培養(yǎng)肝素化的血細胞以刺激非周期的淋巴細胞,之后用秋水仙素處理使其停止在分裂中期相,然后用低滲培養(yǎng)液處理,使細胞核體積漲大,**后用甲醇/冰乙酸固定細胞并制片,用Giemsa染色或進行染色體G顯帶分析。
染色體分析是產前診斷的主要內容之一。近20余年來,遺傳學家對大量流產兒進行了與遺傳有關的細胞核內染色體的研究,發(fā)現50%~60%的流產兒具有異常染色體,包括數目異常和結構異常。這些染色體異常導致了胚胎發(fā)育的障礙,造成妊娠中斷。臨床上對多次流產、曾生育過先天愚型、在妊娠早期有明顯致畸因素接觸的夫婦,都建議做產前宮內診斷。如可在妊娠15~17周時羊膜囊穿刺進行羊水的化學分析和羊水細胞的染色體分析;妊娠8周時即可進行絨毛膜活檢診斷胚胎的染色體異常,達到早期診斷的目的。通過這些產前診斷方法,可將有遺傳病或先天畸形的胎兒篩查出來,進行選擇性流產,控制多種遺傳病的垂直遺傳,達到生育健康后代的目的。
除產前診斷外,染色體分析更多的是用于腫瘤的細胞遺傳學研究。現代腫瘤細胞遺傳學的一個主要結論是,腫瘤細胞的核型變化是不一致地分布在整個染色體組中,不同的腫瘤與不同的染色體區(qū)、帶有關系。即腫瘤的發(fā)生與非隨機性染色體異常密切相關。截**目前,80%以上的腫瘤特異性細胞遺傳學異常,是在血液系統(tǒng)腫瘤發(fā)現和被研究的,因此細胞遺傳學檢查已成為臨床診斷白血病、判斷預后和觀察治療效果的重要方法學之一。
Ph染色體是**早發(fā)現特異性地與惡性腫瘤有關的異常染色體,早在1960年Nowell和Hungerford就在慢性髓細胞性白血病(CML)中發(fā)現了特征性Ph小體,1972年芝加哥大學**細胞遺傳學家Rowley證實,Ph染色體是由第9號染色體和第22號染色體交互易位所致。其后的分子生物學研究又進一步證實了這兩條染色體易位的結果是22號染色體上的BCR基因與9號染色體上的ABL。基因相融合,后來的動物實驗則表明了BCR–ABL融合基因正是造成CML發(fā)病的原因。由于在90%以上的慢性髓細胞性白血病病人中可以檢測到該異常染色體和融合基因,且其異常染色體的比例和融合基因的數量與治療效果和疾病狀態(tài)密切相關,對該染色體的檢查已作為疑診和已確診CML病人的常規(guī)檢查。
20世紀80年代以來,染色體分析發(fā)現90%急性白血病有核型異常,特別是染色體易位,因而提出了白血病的MIC(形態(tài)學、免疫學、細胞遺傳學)分型方法。目前認為急性髓性白血病(AML)中較常見的依次為t(8;21)、[(15;7)、inv(16)/del(16)、t(9;22)等,尤其發(fā)現85%左右M2b存在t(8;21),90%以上急性早幼粒細胞白血病(APL)有t(15;17)易位。從染色體易位的斷裂點已經分離出確切的融合基因,如t(8;21)的AML/ETO融合基因,t(15;17)的PML-RARa融合基因,t(9;22)的BCR–ABL融合基因,以及與11號染色體有關的MLL等。
(3)疾病預后:細胞培養(yǎng)在診斷疾病的預后也具有一定的作用。如重癥再生障礙性貧血的病人,其骨髓集落培養(yǎng),BFU–E的減少與病情嚴重程度是一致的;AML病人的在用藥物后緩解后,如果骨髓的CFU–GM培養(yǎng)檢測,原本減少的集落又恢復生長,而當復發(fā)前集落又減少,所以對估計預防復發(fā)也有一定的意義。
細胞遺傳學分析提供了一些非常有用的預后信息。AML中預后較好的細胞遺傳學異常包括t(8;21)、inv(16)、t(15;17)。如有特征性的染色體5q,7q缺失或單倍體3號染色體的易位或者倒位,t(6;9)、t(9;22)及染色體11q23異常,均提示AML病人預后差。急性淋巴細胞白血病(ALL)中Ph染色體陽性者占約30%,預后不良。
二、細胞培養(yǎng)在臨床治療中的應用
運用細胞進行疾病的治療是20世紀醫(yī)學的一大進步,它使得一些傳統(tǒng)的治療方法束手無策的疾病得到改善,甚**治愈。這種療法稱體細胞治療,它是指應用人體、異體或異體(非人體)的體細胞,經體外操作后回輸(或植入)到人體的療法。傳統(tǒng)的全血輸注、成分輸血、自體和異基因骨髓移植、臍血移植等均屬于體細胞治療的范圍。這些方法主要涉及細胞的采集、保存和運輸。
體細胞治療的類型可以分為三類:①細胞的輸入和植入,旨在體內釋放某些因子,如酶、細胞因子、凝血因子等。②輸入激活的淋巴細胞,如淋巴因子激活的殺傷細胞(LAK)或腫瘤浸潤淋巴細胞(TIL),以及其他能殺傷腫瘤的細胞。③植入經過體外操作的細胞群,如肝細胞、肌細胞、干細胞、胰島細胞等,以求其在體內發(fā)揮復合生物活性作用。干細胞的研究與應用被美G《Science》雜志為1999年世界十大科技成果之一,取自人胚胎或骨髓的干細胞可用于培育不同的人體細胞、組織或器官,這有望成為移植器官的新來源。組織器官移植有可能成為21世紀人類攻克某些重大疾患(如心腦疾病、血液系統(tǒng)疾病等)的根本措施。#p#分頁標題#e#
下面介紹一些臨床常用的細胞培養(yǎng)和擴增。
1.造血祖細胞體外培養(yǎng)和擴增 造血干細胞自我更新的性能使其復制方式為不對稱分裂,且缺乏可靠的人類干細胞定量檢測方法,因此,目前還難以對造血干細胞的擴增進行準確評估,但CD34+造血祖細胞具有良好的擴增反應。這種擴增具有很好的臨床應用前景,一方面可以解決常規(guī)異體移植的配型以及移植物抗宿主反應,另一方面由于采用少量病人的血液,可以避免自身腫瘤細胞污染、供血量不足等問題。
取骨髓、動員外周血、臍帶血,用淋巴細胞分離液分離的單個核細胞,用20%馬血清MEM培養(yǎng)基制成單細胞懸液,應用流式細胞儀進行CD34+細胞的篩選。CD34+細胞在SCF、IL-1、IL-3、IL-5、GM-CSF、C-CSF、EPO、PIXY321等細胞因子的不同組合刺激下,經8~14天即可擴增細胞總數30~1000倍,集落形成細胞(包括CFU-GM、BFU-E、CFU-GEMN)也可被擴增41~190倍。但這些因子的組合在擴增造血細胞的同時,也明顯地加速了造血干/祖細胞的分化,**21天時,jue大部分細胞已分化為較成熟的血細胞,因此,如何在擴增造血細胞的同時,又盡可能地保留造血干細胞并擴增早期造血祖細胞,使其在數量上和功能上滿足臨床治療的需要,這就成為體外擴增研究的重點。近年來,經過各G學者的不斷努力,此方面的研究已取得了明顯的進展,已經有報道,白血病人經過大劑量化療后,應用其外周血擴增的細胞可以重建病人的造血系統(tǒng)。
2.造血祖細胞定向誘導分化 擴增造血祖細胞的同時,也明顯的加速了其分化,這是造血祖細胞擴增所面臨的一個難題,但同時又為我們展開了一個新的研究*域,即利用造血干/祖細胞具有多向分化的潛能,通過細胞因子的不同組合,定向的誘導其分化,產生大量所需的功能細胞(如紅細胞、粒細胞、血小板、樹突狀細胞、NK細胞、淋巴細胞等),滿足基礎研究及臨床應用的需要,這在新一代的細胞免疫治療中將具有十分重要的意義。
(1) 粒系和紅系細胞的定向誘導分化:裴雪濤等利用SCF+IL-3+G-CSF+GM-CSF/或TPO的組合誘導CD34+細胞向粒系分化,5~7天后體系中的粒細胞生長呈現優(yōu)勢,10天時CFU-GM增加了12.87~14.46倍,且出現大量的中性粒細胞;而SCF+ IL-3+EPO+TPO則可擴增BFU-E 15倍,同時體系中出現大量血紅蛋白分泌及網織紅細胞。
(2) 巨核細胞和血小板的定向誘導分化:TPO對巨核系造血祖細胞的增殖分化、血小板的生成及系特異性標志的表達均具有明顯的刺激作用。裴雪濤等利用因子SCF+IL-3+IL-6+TPO進行CFU-MK和細胞CD4la+擴增,于第14天達17~35倍。此外,TPO還可加速CFU–MK進一步分化成熟,誘導巨核細胞在胞漿內形成凸出的界膜系統(tǒng)、血小板特異性顆粒及“血小板區(qū)”,**終分裂形成血小板。
(3) 樹突狀細胞(DC)的定向誘導擴增:DC是體內**有效的抗原提呈細胞(APC)之一,通過其表面的B7、MHC–Ⅱ類分子、ICMC–1等共刺激分子,激活T輔助細胞,促使其分泌IL-2等細胞因子,并傳遞特異性抗原信號,**終激活細胞毒T細胞(CTL)。因此,在腫瘤免疫及其治療中具有重要意義。
(4) LAK細胞、NK細胞和淋巴細胞的定向誘導分化:單用IL-2即可在6~8天誘導CD34+細胞形成NK細胞,CD3-CD56+NK細胞可擴增3~5.4倍,誘導擴增的NK細胞可誘發(fā)移植物抗腫瘤效應(GVT),且對CD34+細胞無明顯影響,SCF、IL-3、IL-15等細胞因子對NK細胞的誘導擴增具有協同作用。目前在臨床上,外周血白細胞經IL-2誘導轉化為LAK細胞,已經成為繼手術、放療、化療后的又一種腫瘤治療方法。
(5) 其他細胞的定向誘導分化:造血干細胞以及更早期的間質干細胞可在體外被誘導分化為造血基質細胞、骨、肌肉、軟骨、肌腱、韌帶等細胞和組織,從而使新一代細胞治療的范圍更加廣闊。
3.自體細胞因子誘導的殺傷細胞 G內陸道培等報道應用自體細胞因子誘導的殺傷細胞(CIK)治療白血病37例,58例次CIK治療,及部分白血病合并丙型病毒性肝炎的效果。采用血細胞分離機大量采集患者的外周血單個核細胞,再用抗CD3單杭、白細胞介素2、干擾素γ培養(yǎng)l0天左右,然后將細胞洗滌后經靜脈回輸給患者。認為自體CIK細胞治療具有明顯清除微小殘留白血病細胞、防止復發(fā)的作用,靜脈輸注安全。并且該治療具有明顯抑制甚**清除白血病患者合并丙型肝炎病毒感染的作用,可明顯改善患者的肝功能。
4.腫瘤細胞疫苗 通過體外細胞培養(yǎng)獲得腫瘤疫苗一直是人們期待的一種腫瘤主動免疫治療手段。雖然目前仍然多在試驗階段,其中一些已展示出臨床應用的樂觀前景。如回輸患者自身的在體外經基因修飾的腫瘤細胞,小鼠中的實驗表明轉染G–CSF的腫瘤細胞可以誘發(fā)機體對腫瘤細胞的特異性細胞免疫。近年來對樹突狀細胞(DC)作為腫瘤疫苗的研究正如火如荼,DC與腫瘤細胞相關抗原、腫瘤細胞裂解物或腫瘤抗原肽(包括合成肽)體外共同孵育,負載了腫瘤抗原的“抗原肽DC瘤苗”、轉基因DC瘤苗、DC與腫瘤細胞的融合瘤苗等均在動物實驗和人體外試驗中顯示了良好抗腫瘤主動免疫效果。
5.皮膚細胞培養(yǎng) 在皮膚受到嚴創(chuàng)傷、燒傷、燙傷時,大面積深度皮膚缺損創(chuàng)面不能自行修復,需要進行皮膚移植。當自體皮膚不能滿足需要時,需要用異體皮膚和人工皮膚。異體皮膚由于排斥反應,一般經2~3周后即發(fā)生溶解脫落。人工皮膚是采用組織工程學和細胞生物學的原理與方法,在體外重組的具有生物學活性的皮膚類似物,按組成成分不同可分為單純人工真皮和具有表皮細胞層的活性復合皮。目前,已研制成功多種人工真皮,如來源于異體或異種(豬)皮的無細胞真皮、以膠原為主要原料經冷凍干燥后形成的海綿狀膠原膜,還有透明質酸膜、聚乳酸/聚羥基乙酸網狀膜等。它們的基本特點是抗原性弱,生物親和性好,植入創(chuàng)而后降解慢,可誘導自體的成纖維細胞、血管內皮細胞及新生毛細血管浸潤生長,形成新的、膠原纖維排列規(guī)則的真皮樣組織,從而重建真皮層。復合皮則是以各種人工真皮為載體,取病人很小的一塊皮膚,分離其中的表皮細胞,在培養(yǎng)箱中培養(yǎng),不斷進行傳代擴增,將擴增的細胞象播種一樣接種在人工真皮上,再培養(yǎng)一段時間,就可以形成含4~6表皮細胞層和真皮層的夾心漢堡包式的人工皮。從理論上分析,取4~20cm#p#分頁標題#e#2自體皮片的表皮細胞于體外培養(yǎng)2~3星期,可形成1000~10 000cm2的人工皮膚。由于復合皮同時含有表皮與真皮層,從而避免了單純表皮細胞膜片移植不耐磨、易收縮、存活率低的缺陷,又避免了單純人工真皮移植仍需移植自體薄皮片的不足。所以,從結構與功能上看,復合皮構成了相對完整的皮膚。所以從嚴格意義上來說,復合皮才是名正言順的人工皮膚。除燒傷外,在大面積的皮膚撕脫傷、瘢痕、慢性難愈性皮膚潰瘍、面部軟組織凹陷等治療中,人工真皮也能發(fā)揮較好的作用,使創(chuàng)面修復質量明顯提高。復合皮的應用可望為大面積皮膚損傷病人提供充足的皮源,并改善創(chuàng)面愈合質量。
6.試管嬰兒 由于培養(yǎng)技術和胚胎學研究的發(fā)展,生殖醫(yī)學理論及其臨床高科技技術也取得了長足進步。出現了試管嬰兒技術,這不僅給治療不孕增添了新的方法和手段,而且也使生殖醫(yī)學基礎理論和生命科學研究進入一個嶄新的*域。**代試管嬰兒,是英GEdwards與Steptoe醫(yī)生創(chuàng)立的經體外受精與胚胎移植,簡稱IVF-ET。此技術對婦女輸卵管不通所致的不孕提供了新的治療方法。第二代試管嬰兒,顯微受精技術,它包括透明帶部分切割技術、透明帶受精技術和單精子卵泡漿內顯微注射受精技術。由于前二者的成功率比較低,所以第二代試管嬰兒主要呈指單精子卵泡漿內顯微注射受精技術(ICSl),適用于重度男性因素不孕治療。第三代試管嬰兒,是指胚胎植入前遺傳學診斷(PGD),此技術是在體外受精,當受精卵發(fā)育到6~8細胞期時.采用顯微技術取出1~2個細胞,采用基因分析或FISH技術,進行遺傳病診斷,去除有病的胚胎,植入正常胚胎而發(fā)育。此技術對預防遺傳病優(yōu)生優(yōu)育發(fā)揮重要作用。
提高種植率是試管嬰兒技術的重點,其關鍵在于胚胎培養(yǎng)和囊胚培養(yǎng)技術。
(1) 胚胎培養(yǎng):
1)胚胎培養(yǎng)液;現在,市面上有多種胚胎培養(yǎng)液出售(表13-10),其應用方法和受精率、妊娠率無明顯差異。目前G內應用**普遍的是HTF和IVF系列,其有效期短,到貨后有效期往往只有3~4周,容易造成短缺或浪費。Q-HTF系列有期較長,根據有些中心應用情況看,胚胎碎片較多,但臨床妊娠率并不低。各個生殖中心可以根據實際情況適當搭配應用。
表13-10 常用胚胎培養(yǎng)液
|
名稱 生產公司 有效期 特點 |
|
HTF Irvine Scientific 3個月 不含HSA |
2) 胎培養(yǎng)方法:目前多數中心培養(yǎng)胚胎**第三天移植,采用微滴法胚胎培養(yǎng),也有一些中心認為采用4孔扳胚胎集中培養(yǎng)有助于胚胎生長,每孔內2~3個胚胎一起培養(yǎng)。原石木郎的研究發(fā)現,第三天以前胚胎單個培養(yǎng)較好,而第三天以后胚胎聯合培養(yǎng)有助于囊胚形成。
有的生殖中心采用微滴法,卵裂率89%,第三天有8個細胞胚胎可供移植占74.9%。胚胎培養(yǎng)注意問題:①市面出售的胚胎培養(yǎng)液內含碳酸鹽緩沖液,孵育時松開瓶蓋,可使CO2進入,新瓶液體取出后**好分裝,避免多次單開瓶口;②去除顆粒細胞的當天,用已經平衡好的培養(yǎng)液滴人生長皿(一般用Falconm皿 3001),每皿6~8滴,每滴25μl,覆蓋礦物油約2ml;
③ 每滴內一般放一個胚胎。
(2) 囊胚培養(yǎng):生理情況下,胚胎在輸卵管內受精后發(fā)育**囊胚時進入子宮進一步卵裂、孵育、著床。改變胚胎移植回子宮的時間,被認為是提高胚胎種植率的方法之一。自1987年以來,移植用的胚胎主要是受精后第2~3天的4~8細胞胚胎。其原因是培養(yǎng)液以及囊胚培養(yǎng)條件的限制造成的。近來出現的序貫培養(yǎng)液,為囊胚移植提供了有利條件,使第5天移植成為可能。#p#分頁標題#e#
1) 培養(yǎng)條件:5%CO2、5%O2、90%N2培養(yǎng)系統(tǒng)降低了氧濃度,低氧環(huán)境使氧自由基產物減少,并可破壞或結合培養(yǎng)基中的某些毒素。空氣中20%的氧深度不是胚胎**適宜的環(huán)境,動物實驗提示胚胎在低氧環(huán)境下種植率提高。日前對這一系統(tǒng)是否有利于培養(yǎng)囊胚的形成仍存在爭議,Noda**報道了在低氧環(huán)境下,Ealre’s液中培養(yǎng)的胚胎及胚胎移植結局好轉。對低氧環(huán)境培養(yǎng)體系這一新的手段尚在評價之中。
在囊胚培養(yǎng)中要求培養(yǎng)基滿足胚胎在體外不同發(fā)育階段生長的需要。胚胎卵裂早期需要微量的葡萄糖,胚泡晚期需要大量的葡萄糖,對丙酮酸的需求則相反。抗氧化牛黃酸對胚泡的發(fā)展是有價值的,次黃嘌嶺是有害的。晚期桑椹胚和胚泡階段胚胎需要非必需氨基酸和必需氨基酸,大約在第3天胚胎基因開始活化并轉錄,若沒有上述氨基酸,必將阻滯囊胚的形成。囊胚培養(yǎng)早期(第3天以前的卵裂階段),胚胎放在簡單介質(如G1)中生長,然后,當胚胎基因開始活化后,再將胚胎轉移到合適介質如(G2中)培養(yǎng),其中不僅包括含鹽、能量底物和蛋白質,而且還有氨基酸、維生素和激素。目前囊胚培養(yǎng)基有多種,如G2、P2、Quinn囊胚培養(yǎng)基等。
2) 培養(yǎng)方法:
方法一:共培養(yǎng)法
共培養(yǎng)法是指用單層細胞作為飼養(yǎng)層細胞,與胚胎共培養(yǎng),模擬體內發(fā)育,促進囊胚的發(fā)育。目前,共培養(yǎng)所用的細胞有人輸卵管壺腹部細胞、vero細胞(輸卵管上皮細胞)、胎牛子宮纖維母細胞、牛輸卵管上皮細胞、人子宮內膜細胞、卵丘細胞及卵巢癌細胞。共培養(yǎng)法的動物實驗開始于20世紀80年代早期,到了90年代對人類胚胎的共培養(yǎng)液進行了大量的研究,人類的胚胎輸卵管細胞上進行受精和生長(共培養(yǎng)),能夠使胚胎的活力提高。
共培養(yǎng)對囊胚形成有重要意義,但在IVF*域存在較大爭議,主要有以下幾個原因:①細胞捐贈人可能的疾病傳播以及利用動物細胞的倫理問題;②由于共培養(yǎng)系統(tǒng)較復雜,且作用機制尚未能夠明確和對應用的有效性**今仍有爭議;③由于其培養(yǎng)體系復雜,可能產生對胚胎的污染。近年來,共培養(yǎng)已逐漸被序貫培養(yǎng)所代替。
方法二:序貫培養(yǎng)法
序貫培養(yǎng)法已經形成并取得了較大的發(fā)展,取代聯合培養(yǎng)系統(tǒng)。序貫培養(yǎng)注意到了人體外培養(yǎng)的胚胎再不同時間里對代謝需求各不相同,即移植前胚胎的營養(yǎng)成分需要改變。對不同發(fā)育時期的胚胎用不同的培養(yǎng)基,而每種培養(yǎng)基適合該時期胚胎發(fā)育的營養(yǎng)需要。Gardner發(fā)展了G1.2、c2.2序貫培養(yǎng)基,他們建立了這兩種培養(yǎng)基的序貫培養(yǎng)系統(tǒng)。G內一些生殖中心也使用G1.2、G2.2序貫培養(yǎng)基培養(yǎng)人囊胚,取得了較滿意的結果。
3)囊胚培養(yǎng)步驟(其中前三天為胚胎培養(yǎng)):
①超促排卵,卵子經4~6小時體外培養(yǎng)成熟。
②體外受精,按10萬條/m1密度將精子加入有卵子的培養(yǎng)孔中,18~20小時后,根據原核及第二極體數檢查受精情況。如果是通過ICSI,取卵后2小時左右,用0.5mg/ml透明質酸酶將卵子外的卵丘消化,再用拉制的玻璃細管將卵子外的顆粒細胞去除。檢查卵子的完好及成熟度,對有**極體的核成熟卵子行ICSI。先將精子置于5%的聚乙烯吡咯烷酮中,經尾部制動后,尾端向前吸人微注射針中,轉移**含HEPEs的HTF管中,保持卵子**極體于6點或12點位置,由3點進針,抽吸少量胞漿以確定卵膜穿透后,將精子置于卵胞漿中央。ICSI后,卵子轉移**HTF培養(yǎng)基中培養(yǎng),次日檢查受精情況。
③第3天,觀察胚胎,如果8細胞I~Ⅱ級胚胎數大于3個,胚胎轉入已經平衡過夜的囊胚培養(yǎng)皿內,評級高的胚胎集中培養(yǎng),碎片含量較多或細胞大小不一胚胎單個培養(yǎng)。第3天胚胎未達到上述標準者,當天移植回子宮。
④檢查囊胚形成率并進行評分,選擇移植或冷凍用胚胎。取卵后第5天,評估胚胎質量,確定胚胎能否用于移植和冷凍。如果再第5天,不能形成囊胚,換液后胚胎繼續(xù)培養(yǎng)1天.在第6天評估是否能用于移植和冷凍。遲于第7天上午形成囊胚者不能再繼續(xù)培養(yǎng)。